




El linfoma del manto fue descrito en 1991 y reconocido por la Organización Mundial de la Salud (OMS) como enfermedad en 1994, es un tipo de linfoma de células B no Hodgkin, que se origina de los linfocitos B situados en la zona del manto del ganglio linfático, se caracteriza por la expresión de marcadores de la línea B (CD19, CD20 y CD5), donde CD3, CD10 y CD23 son negativos y hay sobreexpresión de ciclina D1 asociada a la presencia de la translocación t (11; 14)1. El linfoma de células del manto es un tipo poco frecuente de linfoma no hodgkiniano de células B, que representa del 3 al 10% de todos los linfomas no Hodgkin2. Su incidencia en España es muy baja (0,5 por cada 100.000 habitantes y año)3, afectando con mayor frecuencia a varones (2/1) y a mayores de 60 años. Es un linfoma agresivo, con una supervivencia media entre 3-5 años tras el diagnóstico1, pero gracias a los avances terapéuticos y las estrategias intensivas, la supervivencia se ha duplicado en la última década (60% a los 5 años)4.
La afectación gastrointestinal es infrecuente, en un 10-25%; siendo la forma más común la poliposis linfomatosa múltiple, en la que se identifican múltiples pólipos linfoides en intestino grueso y delgado. Suelen afectar a la región ileocecal, aunque pueden presentarse desde el estómago hasta el recto5. Afectan a colon y recto en el 90%, intestino delgado en el 69%, estómago en el 57% y duodeno en el 52%6. Su diagnóstico endoscópico es infrecuente. Hay descritos casos con invasión microscópica y mucosa normal a la exploración4.
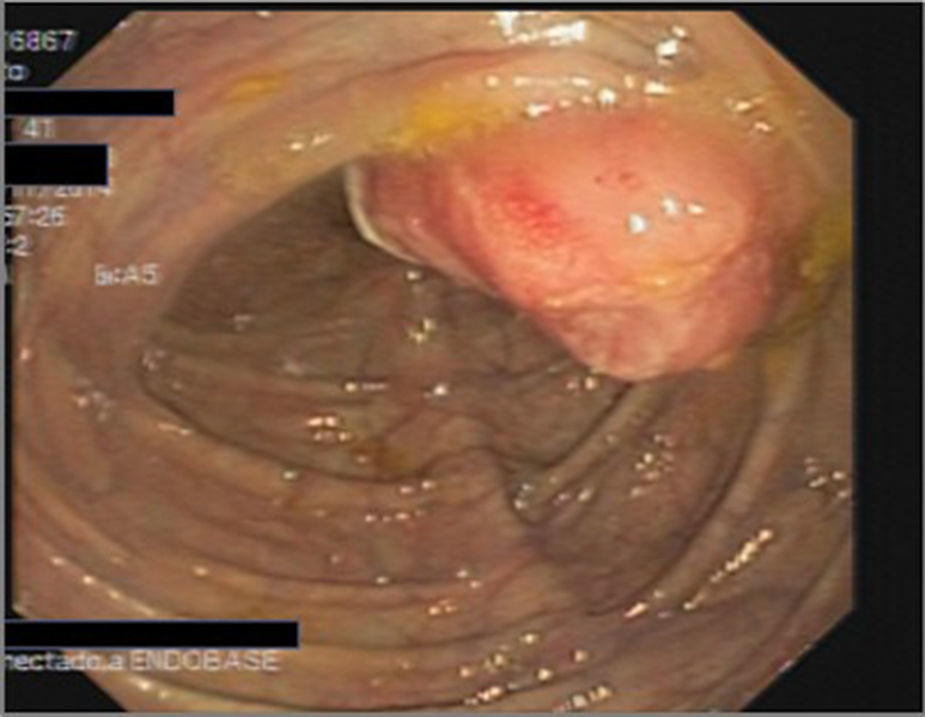
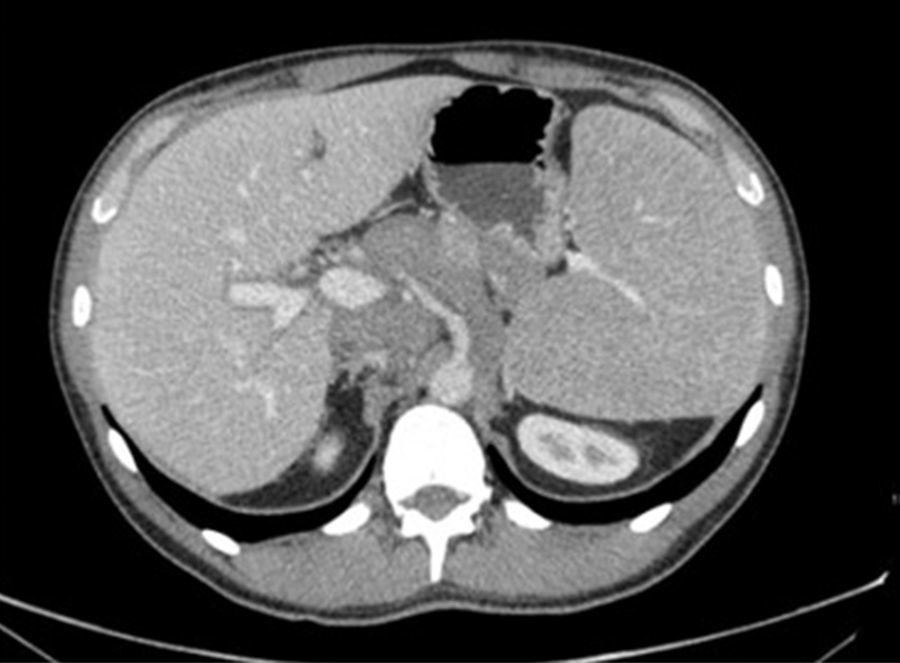
Presentamos el caso de un varón de 41 años, con antecedentes de hipercolesterolemia. Consulta en digestivo por aumento del número de deposiciones y rectorragia de varios días de evolución, sin otra sintomatología acompañante, niega pérdida de peso, dolor abdominal y fiebre. A la exploración física se palpa una esplenomegalia. Analíticamente presenta una marcada elevación de la LDH, siendo el resto de parámetros bioquímicos y del hemograma normales. Se solicita una colonoscopia en la que apreciamos una formación polipoidea sésil (fig. 1) friable y ulcerada, de unos 30-40mm localizada en ciego, de la que se toman múltiples biopsias, no se puede franquear válvula ileoceal a pesar de varios intentos. Con los resultados obtenidos solicitamos una TC toraco-abdominal con contraste iv (fig. 2), que objetiva múltiples adenopatías retropectorales mediastínicas, mesentéricas, retroperitoneales y pélvicas, además de un gran conglomerado/masa que engloba a los vasos del retroperitoneo superior.


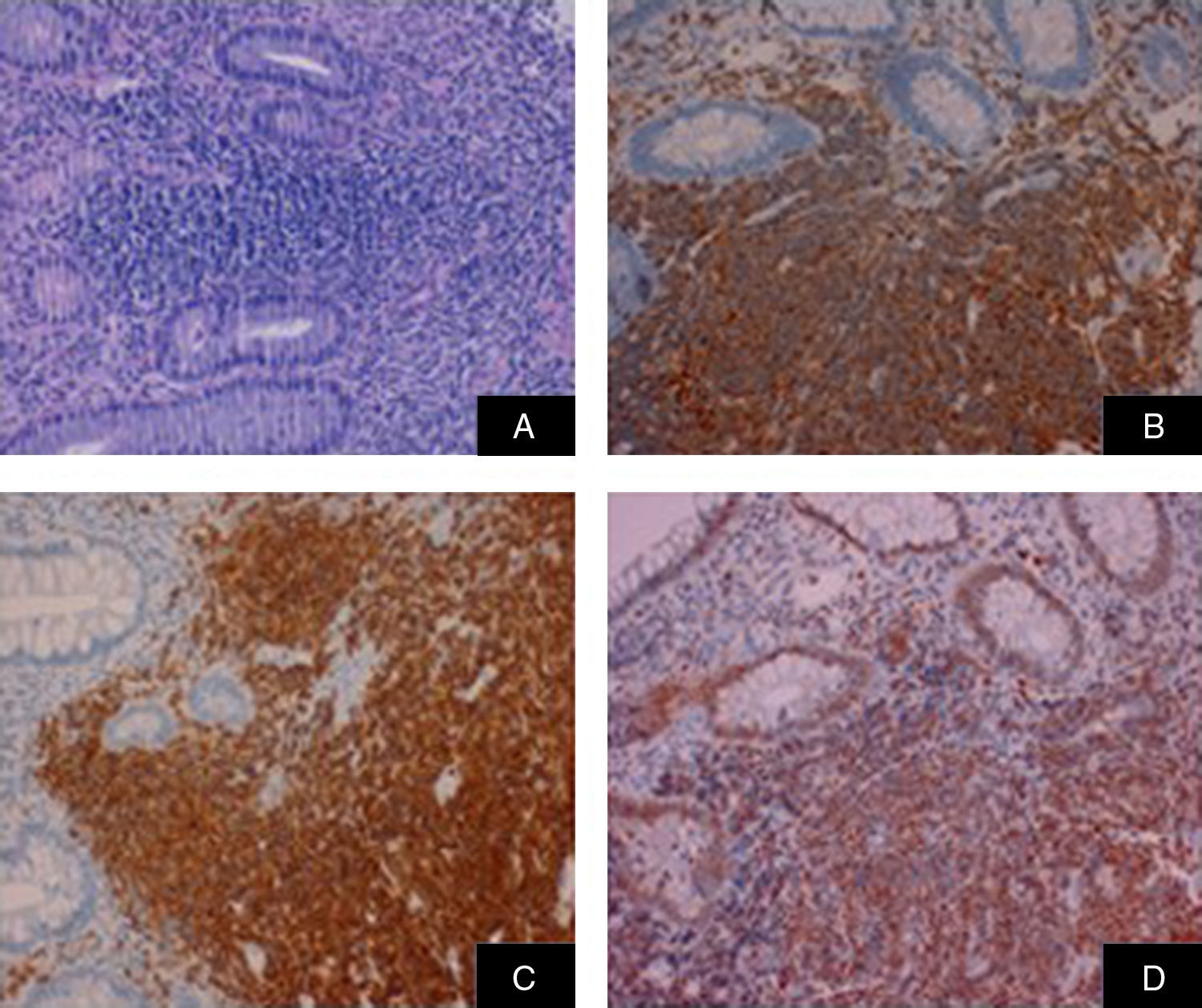
El resultado histológico (fig. 3) de las biopsias endoscópicas muestra una proliferación linfoide atípica con fuerte expresión CD20, CD5 y ciclina D1 en más del 75% de la población de linfocitos B, compatible con infiltración por linfoma no Hodgkin B de células del manto. Se completa el estudio con un PET/TC que evidencia una afectación linfática supra e infra-diafragmática, esplenomegalia linfomatosa, afectación de médula ósea y del colon de forma difusa, siendo compatible con un estadio metabólico IV.
 Población linfoide tumoral. B) Inmunorreacción positiva CD5. C) Positiva CD20. D) Positiva ciclina D1.")
El paciente inicia tratamiento alternante con rituximab-ciclofosfamida, vincristina, prednisona (R-macro CHOP)/rituximab-dexametasona, cisplatino, citarabina (R-DHAP), recibiendo un total de 6 ciclos, con respuesta completa. A los 6 meses del tratamiento, el PET/TC toraco-abdominal de control no evidencia depósitos patológicos del radiotrazador. La gastroscopia y la colonoscopia de revisión no muestran ninguna lesión. Dado que presenta una adecuada respuesta a la terapia intensiva, se realiza trasplante autógeno de sangre periférica (TASP) como consolidación del tratamiento. Un año después del TASP el paciente se mantiene en remisión completa, en tratamiento de mantenimiento con rituximab.
Nuestro paciente es menor de 60 años, con la peculiaridad de no presentar múltiples pólipos, sino un pólipo único de gran tamaño. En el 90% de los pacientes la sintomatología es inespecífica, como: pérdida de peso, astenia, letargia, fatiga, anemia, masa abdominal o rectal palpable y adenopatías palpables. La afectación de la médula ósea se aprecia en estadios avanzados7. Cuando existe afectación gastrointestinal por el linfoma, los síntomas digestivos se han descrito entre el 15-30% de los casos; se recomiendan estudios endoscópicos principalmente si hay dolor abdominal, alteración del hábito intestinal o rectorragia, como sucedió en nuestro caso6.
El linfoma de células del manto se considera un tipo agresivo de linfoma, de progresión rápida. El 80% de los pacientes son diagnosticados en estadios avanzados7. Para mejorar el tratamiento, debe realizarse un pronóstico. Los índices pronósticos permiten desarrollar estrategias de tratamiento en función de los factores de riesgo particulares del paciente. En el linfoma de células del manto se utiliza el índice pronóstico internacional del linfoma del manto (MIPI)8. En pacientes menores de 60 años y/o elevada puntuación en índice MIPI se recomiendan estrategias terapéuticas intensivas con R-CHOP, R-bendamustina y/o R-DHAP, seguido de TASP9. Nuestro paciente presentaba un MIPI de 8 puntos, lo que indica un alto riesgo (supervivencia media estimada de 29 meses), por lo que recibe una estrategia terapéutica intensiva con R-macro CHOP/R-DHAP y consolidación con TASP, manteniéndose remisión completa 30 meses tras el tratamiento quimioterápico inicial.
Presentan un peor pronóstico los pacientes con edad avanzada, estadio avanzado al diagnóstico, albumina baja, esplenomegalia, LDH elevada y anemia9.
Al Servicio de Anatomía Patológica del Hospital Virgen Macarena por proporcionar imágenes completas de la biopsia, y al Servicio de Hematología del Hospital Virgen de Valme por los datos ofrecidos sobre el manejo de estos casos.

