




El síndrome de POEMS1 (polineuropatía, organomegalia, endocrinopatía, proteína monoclonal y cambios en la piel) se caracteriza por la presencia de un trastorno en las células plasmáticas monoclonales, neuropatía periférica y una o más de las siguientes características: mieloma osteosclerótico, enfermedad de Castleman, elevación del factor de crecimiento vascular endotelial (VEGF), organomegalia, endocrinopatía, edema, cambios típicos de la piel y papiledema2. Presentamos el caso de una paciente con polineuropatía asociada a un síndrome de Budd-Chiari resultante de una presentación inusual de síndrome de POEMS.
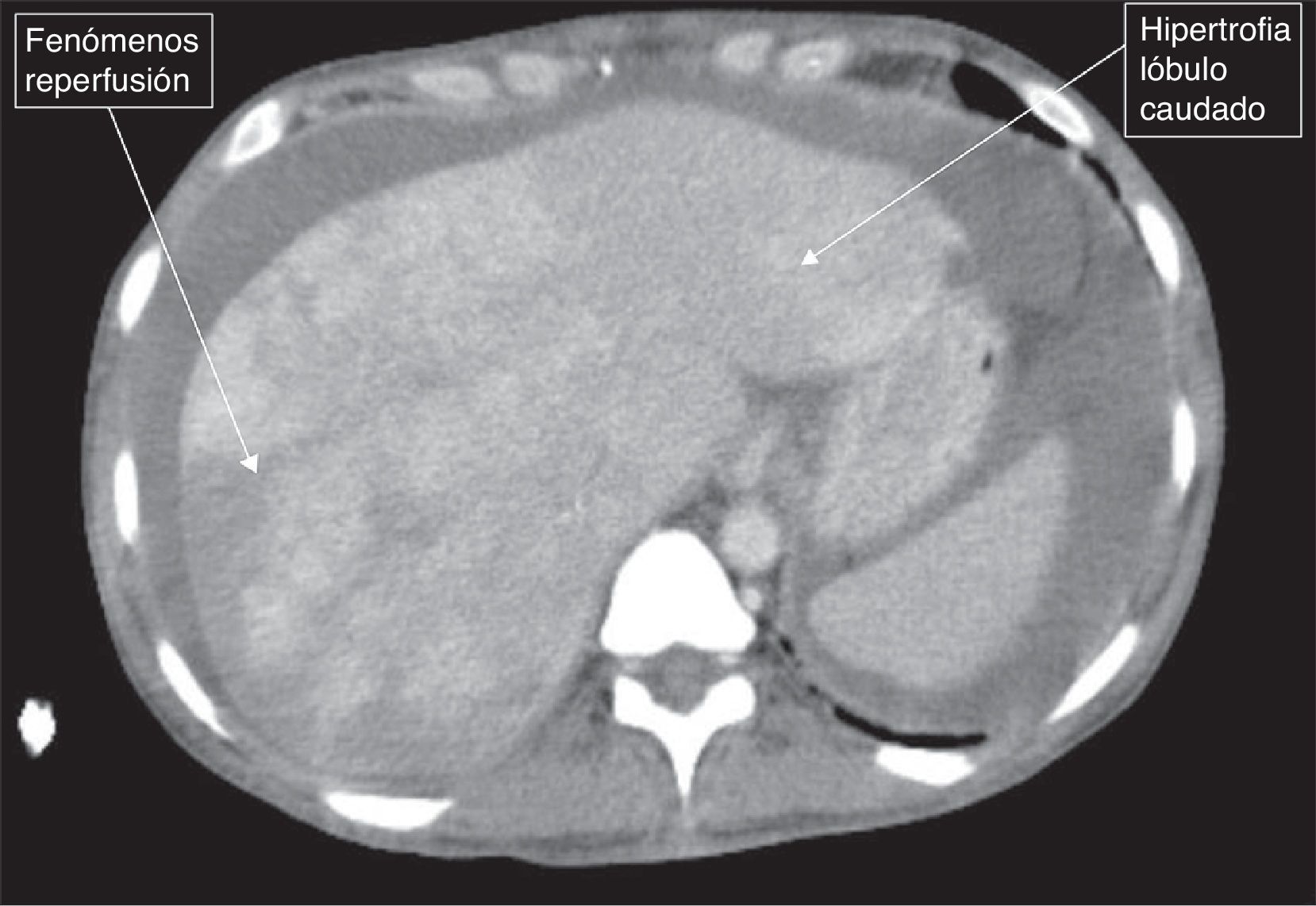
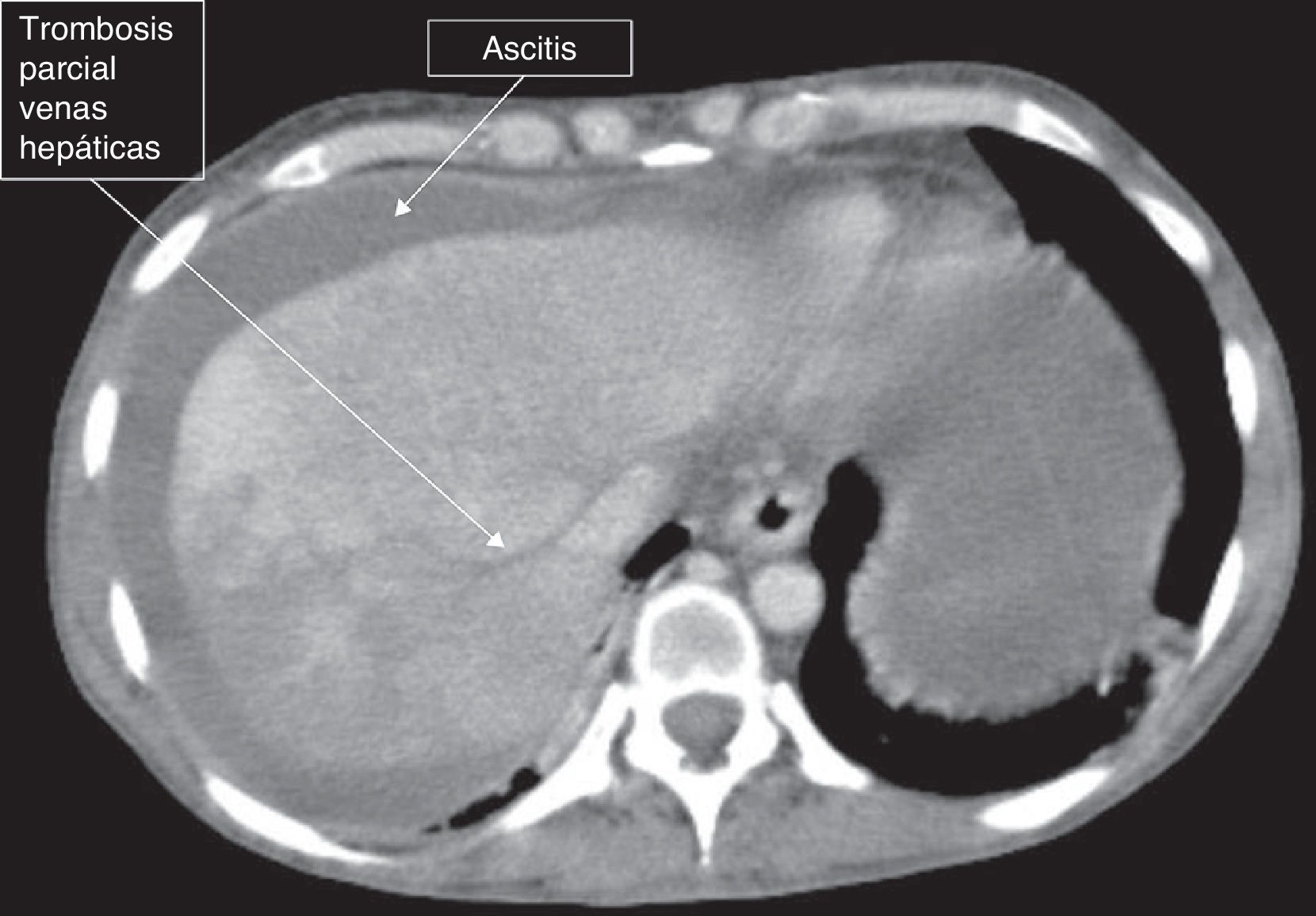
Se trata de una mujer de 44 años de edad, afecta de hipotiroidismo, en estudio por parestesias y debilidad de ambos pies por el que se realizó el diagnóstico de polineuropatía desmielinizante inflamatoria crónica. La exploración física mostró hiperpigmentación de la piel y hepatomegalia. Los análisis séricos fueron normales, salvo la detección de anti-HBs y anti-HBc (IgG). La ecografía reveló esplenomegalia, discreta ascitis y circulación portal y hepática patentes. Dado que presentaba mínima ascitis en este momento no se realizó paracentesis. En una primera evaluación se realizó una medición del gradiente de presión venosa hepática, siendo la presión hepática enclavada de 15,5mmHg, la presión hepática libre de 9,67mmHg, la presión en la cava de 8,39mmHg y la biopsia hepática transyugular normal. Además se realizó estudio ecocardiográfico que no mostró alteraciones de la función cardiaca. Debido a la polineuropatía se inició tratamiento con inmunoglobulina intravenosa. Diez meses más tarde, la ascitis se había incrementado hasta hacerse casi a tensión, y la paciente fue hospitalizada de nuevo. Se realizó angio-TAC hepático objetivando hepatomegalia heterogénea a expensas del lóbulo caudado (fig. 1), ascitis y trombosis parcial de las venas hepáticas (fig. 2). Se calculó el gradiente de albúmina: 1,7 (albúmina sérica 3,1g/dl y albúmina en líquido ascítico 1,4g/dl). Se realizó de nuevo una medición de la presión venosa hepática, siendo la presión ‘enclavada’ de 20,31mmHg y la «libre» de 15,58mmHg, por lo que el gradiente de presión venoso hepático resultó en ese momento normal. Se realizó nueva biopsia hepática, que en esta ocasión reveló dilatación sinusoidal, congestión vascular y necrosis de la zona3. Mediante inmunofijación se detectó una paraproteína monoclonal IgA lambda en suero (1,06g/dl). Posteriormente, la paciente presentó episodios de visión borrosa mostrando el estudio oftalmológico edema de papila bilateral; la RMN craneal evidenció trombosis parcial de la arteria carótida interna derecha y se inició tratamiento anticoagulante. Ante la sospecha diagnóstica de un síndrome de POEMS se procedió a la biopsia de médula ósea y al estudio de la misma mediante citometría de flujo, identificando menos del 2% de células plasmáticas atípicas. Los niveles de VEGF en plasma fueron de 1.951pg/ml. En esos momentos la paciente presentaba: Cr 1,11mg/dl, Na 130mmol/l, bilirrubina 0,69mg/dl, colinesterasa 3.300U/l, albúmina 3.100g/dl, plaquetas 148.000 y Quick 74%. Ante estos hallazgos, la paciente fue diagnosticada del síndrome de POEMS, complicado con un síndrome de Budd-Chiari. Se procedió a tratamiento con melfalán y posterior trasplante de células hematopoyéticas (HCT), realizándose previamente una derivación portosistémica intrahepática (DPPI) e inicio de tratamiento anticoagulante.


La existencia de un episodio trombótico dentro de un síndrome de POEMS no es un episodio frecuente y en nuestro conocimiento apenas se han referido en la literatura pacientes con trombosis de las venas suprahepáticas como manifestación inicial de este síndrome. Por ello el proceso diagnóstico fue difícil, lo que ya ha sido reconocido en general, en las series más extensas como la descrita en la clínica Mayo2. La causa de este raro síndrome es desconocida, aunque una sobreproducción crónica proinflamatoria y de citocinas, son los principales mecanismos implicados4. La prevalencia de hepatomegalia y ascitis es variable según las diferentes series de casos2,5–7. Probablemente, la organomegalia esté en relación al cierto grado de congestión vascular, la cual se atribuye a los niveles elevados de VEGF3. Usualmente la histología hepática en los pocos casos en los que se dispone, muestra la presencia de hiperplasia nodular regenerativa o cirrosis6,8 (en los menos) y tan solo se han publicado en nuestro conocimiento otros 2 casos como nuestra paciente, con síndrome de Budd-Chiari2. Los pacientes con síndrome de POEMS pueden padecer alteraciones trombóticas, siendo los más frecuentes episodios cerebrales y cardiacos2,9. Hay poca información sobre el análisis del líquido ascítico en el síndrome de POEMS, y en los casos en los que se ha mencionado se ha descrito como una ascitis de «bajo gradiente» no relacionado con la presencia de hipertensión portal10. En nuestro caso, sin embargo, el gradiente resultó elevado por lo que se consideró secundario a la presencia del síndrome de Budd-Chiari. Por ello y para llevar a cabo un tratamiento eficaz de la hipertensión portal se decidió la realización de una derivación portal percutánea intrahepática (DPPI). Optamos por la realización de la DPPI debido a que la paciente precisaba de forma «rápida» mejorar la función hepática, previamente al inicio de quimioterapia con melfalán, la cual fue también complementada con un trasplante de células hematopoyéticas. La paciente 3 años después del inicio del cuadro se encuentra en situación de franca mejoría clínica habiendo recuperado gran parte de su déficit neurológico, sin presentar otras alteraciones de la función hepática y siendo la DPPI permeable.
En conclusión, esta carta plantea la necesidad de incluir el síndrome de Budd-Chiari entre las manifestaciones clínicas del raro síndrome de POEMS y describe algunas de las peculiaridades de esta infrecuente asociación.

