






El tratamiento del dolor en el paciente con cirrosis hepática es un verdadero reto, siendo muchas veces inadecuado por falta de eficacia terapéutica o por la gran incidencia de efectos adversos. El enfoque del tratamiento es diferente si el dolor es agudo o crónico e implica conocer el mecanismo fisiopatológico responsable del mismo. El tratamiento farmacológico se ha de iniciar con la dosis mínima efectiva y titular lentamente, evitando la polifarmacia. Se enfatiza el control de los efectos adversos, especialmente la sedación y el estreñimiento que predisponen el desarrollo de la encefalopatía hepática. El paracetamol es el primer eslabón terapéutico siendo seguro a dosis 2-3 g/día. Los antiinflamatorios no esteroideos están contraindicados ya que pueden inducir insuficiencia renal aguda y/o sangrado gastrointestinal. El tramadol es una opción segura en el tratamiento del dolor moderado a severo. Los opioides con mayor seguridad terapéutica son el fentanilo, la hidromorfona y la metadona como segunda opción. El tratamiento tópico puede disminuir el consumo de fármacos por vía oral. En el tratamiento del dolor neuropático la gabapentina es la primera opción terapéutica, mientras que los antidepresivos tricíclicos pueden estar indicados en cierto grupo de pacientes. Las técnicas intervencionistas son una herramienta valiosa a utilizar en el dolor moderado a severo ya que permiten disminuir el tratamiento farmacológico y con ello los efectos adversos. Las intervenciones psicológicas, la terapia física y la rehabilitación deben ser consideradas como parte de la terapia multimodal en el abordaje del dolor crónico.
Pain management in patients with liver cirrhosis is a real challenge and is often inadequate due to a lack of therapeutic efficacy or the high incidence of adverse effects. The focus of treatment differs depending on whether the pain is acute or chronic and involves understanding the causative pathophysiological mechanism. Analgesics should be started with the minimum effective dose and should be titrated slowly with avoidance of polypharmacy. Adverse effects must be monitored, especially sedation and constipation, which predispose the patient to the development of hepatic encephalopathy. The first-line drug is paracetamol, which is safe at doses of 2-3g/day.
Non-steroidal anti-inflammatory agents are contraindicated because they can cause acute renal failure and/or gastrointestinal bleeding. Tramadol is a safe option for moderate-severe pain. The opioids with the best safety profile are fentanyl and hydromorphone, with methadone as an alternative. Topical treatment can reduce oral drug consumption. In neuropathic pain the first-line therapeutic option is gabapentin. The use of antidepressants such as amitriptyline can be considered in some patients. Interventional techniques are a valuable tool in moderate to severe pain, since they allow a reduction in drug therapy and consequently its adverse effects. Psychological treatment, physical therapy and rehabilitation should be considered as part of multimodality therapy in the management of chronic pain.
La cirrosis hepática (CH) es la consecuencia final de distintas enfermedades hepáticas crónicas que lleva a la pérdida de la arquitectura normal del hígado con disminución progresiva de sus funciones, y se define por la presencia de cambios anatómicos en el parénquima hepático que incluyen la fibrosis y el desarrollo de nódulos de regeneración1. Es un importante problema de salud pública y conlleva una elevada morbilidad y mortalidad2. El dolor, tanto agudo como crónico, es un síntoma que se presenta con mucha frecuencia en estos pacientes3. A pesar de que en los últimos años se han hecho importantes avances en el diagnóstico, seguimiento y tratamiento de la CH1, el tratamiento del dolor sigue siendo un verdadero reto.
La prevalencia del dolor en los pacientes con enfermedad hepática es elevada, oscila entre un 30 y un 40%3. Estos pacientes, además de presentar las mismas causas de dolor que la población general, pueden presentar dolor relacionado con la enfermedad. Es frecuente el dolor abdominal o lumbar asociado a la ascitis y la mastalgia o dolor torácico asociado a ginecomastia3,4. Además, son más propensos a sufrir fracturas por la elevada incidencia de osteoporosis5, y la inmunodepresión conlleva que entidades como el herpes zóster y la neuralgia postherpética (NPH) no sean infrecuentes.
En muchas ocasiones, el tratamiento del dolor en los pacientes con CH es inadecuado (tabla 1), bien sea por una terapéutica poco efectiva o por la elevada incidencia de efectos adversos. La mayoría de los fármacos analgésicos son metabolizados en el hígado por lo que estos pacientes son susceptibles de sufrir complicaciones, que con frecuencia son graves y pueden comprometer la vida del paciente, como la encefalopatía hepática (EH), el sangrado gastrointestinal (GI) o la insuficiencia renal (IR)4,6,7.
Causas del tratamiento analgésico inadecuado en el paciente con enfermedad hepática avanzada
| 1. Falta de asociación entre la clasificación clínica de la severidad de la cirrosis hepática y el grado de afectación del metabolismo del fármaco |
| 2. Ausencia marcador analítico para valorar el grado de afectación del metabolismo |
| 3. Escasos ensayos clínicos con correcta evidencia científica |
| 4. Falta de guías clínicas basadas en la evidencia |
| 5. Enfermedades asociadas como la enfermedad renal |
| 6. Polifarmacia |
| 7. Reacciones adversas conllevan la escasa adherencia al tratamiento |
| 8. Miedo, inexperiencia o falta de conocimientos del personal sanitario |
Dentro de las múltiples causas del tratamiento inadecuado destacan la falta de asociación que existe entre la clasificación clínica de la severidad de la cirrosis con el grado de afectación en el metabolismo de los fármacos3,4,6–10. Otro punto destacable es que no hay un marcador analítico reproducible en la clínica que pueda utilizarse para medir el metabolismo hepático de los fármacos o un parámetro capaz de medir de manera certera la función residual hepática para poder ajustar la dosis3,4,8,10. La falta de ensayos clínicos, metodológicamente bien elaborados, que analicen el tratamiento analgésico y su correlación con los efectos adversos y de guías de práctica clínica consensuadas no facilita el diseño de tratamiento adecuado4,6,7,10,11. A todo lo anterior se añade el complejo manejo per se de estos pacientes, donde es frecuente tanto la alteración de la función renal (asociada o no a la enfermedad hepática), como la polifarmacia (enfermedades crónicas concomitantes) lo que los hace más vulnerables a la aparición de interacciones farmacológicas. Por último, probablemente la causa más común de tratamiento inadecuado se deba al miedo, inexperiencia o falta de conocimientos por parte del personal médico.
Comportamiento farmacológico de los analgésicos en el paciente con enfermedad hepática avanzadaEl metabolismo hepático de los fármacos se inicia con la llegada del fármaco al hígado, para ser captado por los hepatocitos donde se va a metabolizar siguiendo reacciones de oxidación, hidrólisis, hidroxilación, reducción o demetilación (reacción de fase i) y/o reacciones de conjugación a ácido glucurónico, acetato, glicina, etc. (reacción de fase ii)7. Depende del flujo sanguíneo hepático, de la afinidad a la unión con las proteínas plasmáticas7 y de la capacidad de metabolización de las enzimas hepáticas. Otras situaciones como la presencia de cortocircuito portosistémico y/o enfermedad renal asociada (síndrome hepatorrenal [SHR]) pueden alterar aún más la función metabólica (tabla 2). Los pacientes con enfermedad hepática crónica compensada tienen un metabolismo de los fármacos similar a la población sana. En pacientes con disfunción hepática por colestasis y/o hepatitis, y los pacientes con cirrosis compensada tienen un mejor metabolismo de los fármacos que los pacientes con cirrosis evolucionada e hipertensión portal (HTP)6,7. Mientras mayor es la disfunción hepática, peor es la capacidad del hígado para metabolizar los medicamentos.
Causas que modifican el metabolismo hepático de los fármacos en el paciente con enfermedad hepática avanzada
| 1. Necrosis de los hepatocitos |
| 2. Cortocircuito portosistémico |
| 3. Disminución de la cantidad del fármaco unido a proteínas plasmáticas |
| 4. Volumen de distribución anormal |
| 5. Alteración en la eliminación del fármaco |
| 6. Alteración per se del metabolismo del fármaco |
| 7. Alteración farmacodinámica |
| 8. Enfermedad renal asociada |
| 9. Interacciones farmacológicas |
Los fármacos con gran metabolismo de primer paso (alta extracción hepática) tienen mayor cantidad de fármaco libre con aumento de la biodisponibilidad (BD), agravándose con la presencia de hipoproteinemia. Es importante considerar que el metabolismo de determinados analgésicos, especialmente los opioides, generan metabolitos activos y que la capacidad para eliminarlos puede estar alterada en estos pacientes. Todas estas consideraciones definen qué fármacos son seguros, la necesidad de reducir la dosificación y el régimen de prescripción6,7,9.
El estado nutricional, la función renal, el uso/abuso de alcohol, las interacciones farmacológicas o si es candidato a trasplante hepático son otras situaciones que se deben considerar. En tratamientos de larga duración la monitorización de la función hepática y de los efectos secundarios y, si es necesario, medir los niveles plasmáticos del fármaco6,7.
Dolor en el paciente con cirrosis hepáticaAntes de adentrarnos en el tratamiento del dolor se hace necesario recordar ciertas definiciones. El dolor se define como una experiencia sensorial y emocional desagradable, asociada con un daño tisular, real o potencial, o descrita en términos de dicho daño12. De esta definición se extrae que el dolor no solo comprende la respuesta biológica que ocurre como consecuencia de la interacción de un estímulo lesivo con nuestro organismo sino que involucra parámetros emocionales independientes para cada persona que están influidos por el entorno socioeconómico, el genotipo individual, las conductas previamente aprendidas o la personalidad. Por esta razón, la experiencia dolorosa es siempre subjetiva e independiente para cada individuo.
El dolor se puede clasificar desde el punto de vista temporal en dolor agudo y dolor crónico. El dolor agudo es considerado como un mecanismo de defensa ante un suceso perjudicial, y normalmente desaparece cuando la causa del dolor se ha resuelto, mientras que el dolor crónico es el que persiste más allá de la lesión causal que lo originó. De manera arbitraria se ha establecido que ha de durar más de 3 a 6 meses y se considera en sí mismo una enfermedad12. Para el tratamiento adecuado del dolor se ha de entender el mecanismo fisiopatológico responsable del mismo13–15. Simplificando, el dolor nociceptivo se produce cuando un estímulo activa los nociceptores localizados en los tejidos externos (dolor somático) o internos (dolor visceral); esta señal viaja a través del sistema nervioso y termina con la interpretación de la misma en el cerebro. El dolor neuropático (DN) se define como el dolor causado por lesión o enfermedad del sistema nervioso somatosensorial12, tiene diferentes etiologías y localizaciones, siendo las causas más comunes las radiculopatías, la polineuropatía diabética (PND) y el traumatismo nervioso que incluye el dolor posquirúrgico16,17.
Parece evidente que la incidencia de dolor está asociada con la severidad de la enfermedad hepática aunque no está claro que esté relacionado con la etiología de la enfermedad hepática. Sin embargo, la fibromialgia y la dispepsia asociadas con depresión tienen prevalencia en pacientes con CH por virus C. En la hepatopatía alcohólica la presencia de polineuropatía es muy relevante3.
La mayoría de los pacientes se ven beneficiados por un manejo multidisciplinario acompañado de distintas modalidades terapéuticas (terapia intervencionista, fisioterapia, etc.)13,14,18. Los pacientes con dolor crónico presentan con frecuencia ansiedad, depresión y/o trastornos del sueño que deben ser tratados12,14. La terapia farmacológica multimodal es la más aceptada ya que los distintos tratamientos actúan de manera diferente y a distintos niveles de la vía de transmisión del dolor13–15. Siempre que sea posible hay que tratar la causa del dolor y cuando existe dolor intenso que no mejora o hay empeoramiento del dolor inicial se han de investigar las causas potencialmente tratables, o implementar otras medidas (terapia física, bloqueos nerviosos) antes de incrementar la dosis de los analgésicos13.
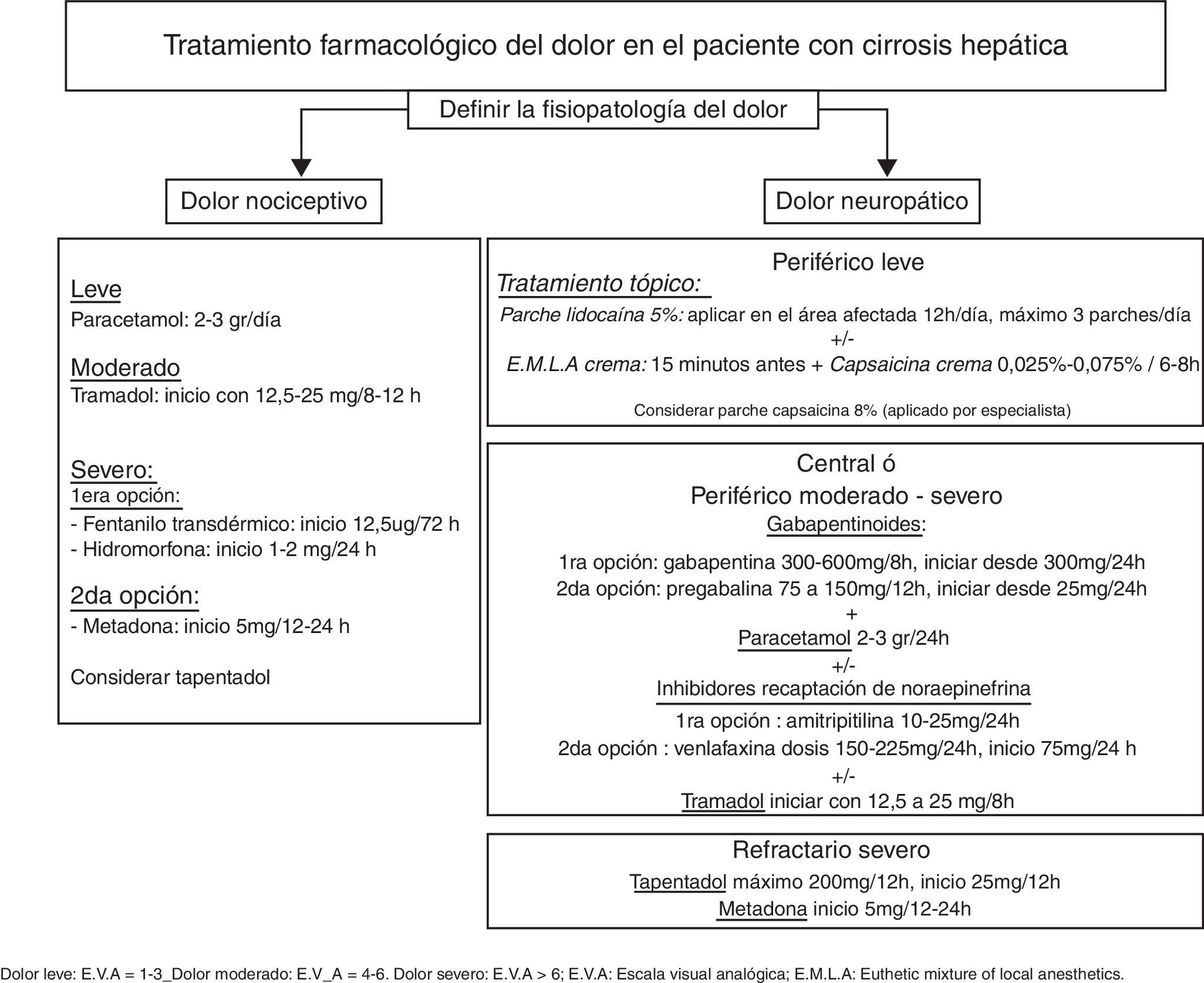
Tratamiento farmacológicoTodos los fármacos tienen efectos adversos y se deben utilizar solo si los beneficios superan los riesgos, respetando las expectativas, experiencias y preferencias del paciente13. El médico responsable de iniciar el tratamiento debe valorar toda la medicación del paciente, el estado nutricional, la función renal, el consumo de alcohol o si es candidato a un trasplante hepático6 evitando potenciales interacciones farmacológicas y aparición de efectos adversos4. Se han de dosificar los analgésicos lentamente, comenzando con la menor dosis recomendada, monitorizar continuamente la aparición de efectos adversos y evitar la polifarmacia. En la figura 1 se presenta un esquema de tratamiento farmacológico del dolor en el paciente con CH.
Paracetamol (Acetaminofén)
Es un analgésico y antipirético sin efectos antiinflamatorios con un excelente perfil de seguridad en todos los grupos etarios19, por lo que muchas sociedades científicas lo consideran el primer fármaco que se debe utilizar en pacientes con dolor musculoesquelético crónico20,21. Su mecanismo de acción sigue siendo tema de debate; la hipótesis más aceptada es que actúa en el sistema serotoninérgico del sistema nervioso central (SNC) y aunque probablemente exista una actividad sobre las ciclooxigenasas, esta sería diferente a la de los antiinflamatorios no esteroideos (AINE)22. No produce efectos adversos importantes gastrointestinales y no inhibe la actividad plaquetaria, lo que hace que sea una opción óptima para los pacientes con riesgo de sangrado gastrointestinal19. Tiene metabolismo hepático y sus metabolitos son responsables de la hepatotoxicidad observada en la sobredosis19 (hepatitis fulminante). Existe la idea en muchos profesionales de salud que el paracetamol debe evitarse en pacientes con enfermedad hepática, y recomendando el uso de AINE en detrimento de este fármaco, sin valorar el grado de perjuicio que esto supone4.
Hay evidencia de toxicidad hepática con dosis terapéuticas de paracetamol en pacientes con enfermedad hepática. Un porcentaje relativamente pequeño de este fármaco se metaboliza por vía del citocromo P450 a benzoquinona, muy hepatotóxica, que mediante conjugación con el glutatión hepático se transforma en metabolitos no tóxicos que se eliminan por vía renal. La ingesta de alcohol, por depleción de la reserva hepática de glutatión y niveles bajos de albúmina debida a desnutrición, son responsables del aumento de benzoquinona y consecuente necrosis hepatocelular4,19,23.
La hepatotoxicidad clínica en pacientes sanos es extraordinariamente rara con dosis menores a 4g/día. No hay estudios prospectivos de largo seguimiento que evalúen la seguridad del paracetamol en los pacientes cirróticos4,6, aunque existe algún reporte que corrobora la no asociación entre consumo de dosis bajas de paracetamol y la hospitalización por complicaciones hepáticas24. La recomendación de expertos para uso crónico del paracetamol (> 14 días) en pacientes con CH es 2-3g/día, mientras que para uso agudo 3-4g/día parece seguro4,6. Los pacientes malnutridos o que consumen alcohol de forma crónica son población de riesgo y la recomendación es dar menos de 2g/día4.
Antiinflamatorios no esteroideosLos AINE son los fármacos más utilizados para el tratamiento del dolor crónico no oncológico, a pesar de que muchas guías de tratamiento recomiendan que su uso deba ser minimizado, sobre todo en pacientes geriátricos20,21. Actúan inhibiendo la enzima ciclooxigenasa (COX) que es la encargada de la conversión del ácido araquidónico en prostaglandinas (PG) y tromboxanos. Esta inhibición es responsable de los efectos farmacológicos deseados (antiinflamatorio, analgésico y antipirético) y de los efectos adversos. Son metabolizados fundamentalmente por el sistema del citocromo P450 y tienen amplia unión a proteínas plasmáticas, lo que condiciona un aumento de los niveles plasmáticos en los pacientes cirróticos4,6. Pueden inducir hepatotoxicidad por necrosis hepatocelular, colestasis aguda o crónica e idiosincrásica6. La tasa de filtración glomerular y el flujo sanguíneo renal dependen del efecto vasodilatador de las PG en los pacientes con enfermedad hepática avanzada, por lo que la disminución de los niveles de estos mediadores inflamatorios ocasionan descenso de la perfusión renal, reducción del filtrado glomerular y aumento de la retención de sodio6. Asimismo, el sangrado gastrointestinal es más frecuente en este grupo de pacientes, siendo mayor en pacientes con HTP y gastropatía y varices esofágicas4,6. Por todo ello, están contraindicados, incluso en pacientes con CH compensada4,6,7.
Con respecto a los AINE COX-2 selectivos no hay estudios que evalúen su seguridad en el paciente con CH. A pesar de que tienen menos efectos adversos gastrointestinales o plaquetarios cuando se compara con los AINE no selectivos, tienen importantes efectos deletéreos a nivel cardiovascular o renal, ya que la COX-2 se expresa de manera constitutiva en la corteza renal y está vinculada a la mediación de la liberación de renina, la regulación del sodio y el mantenimiento del flujo sanguíneo renal25, lo que justifica su prescripción restrictiva en los pacientes hepatópatas. El meloxicam se ha vinculado con la aparición de hepatitis autoinmune26.
Entre los gastroenterólogos el Metamizol (dipirona) es un analgésico que goza de mayor aceptación. Es buen antipirético y analgésico aunque es menos antiinflamatorio que otros AINE. Su acción analgésica es por mecanismo periférico y centrales a nivel medular y SNC. Activa la vía óxido nítrico-GMP cíclico y los canales de potasio a nivel periférico, lo que explica su efecto antiespasmódico27. Dentro de sus efectos adversos destaca la agranulocitosis y la aplasia de médula ósea idiosincrásica, por lo que, aunque hay estudios que informan de la baja incidencia28, su uso se ha prohibido en varios países. La inhibición de la síntesis de PG es menos potente que con otros AINE por lo que se cree que es mejor tolerado aunque no hay evidencia que soporte este hecho. Induce las enzimas del citocromo P450 por lo que puede causar múltiples interacciones farmacológicas. Está contraindicado en el paciente cirrótico y si se administra se debe ajustar la dosis y el tiempo de administración29.
OpioidesSon los analgésicos más utilizados en el tratamiento del dolor moderado y severo30,31. Su uso en pacientes con dolor crónico no oncológico sigue siendo tema de debate, aunque pueden prescribirse cuando el resto de tratamientos son inefectivos y/o están contraindicados31. Históricamente se han utilizado en el tratamiento del dolor severo, aunque no todos los tipos de dolor responden a estos fármacos, y muchos de los pacientes que se benefician de este tratamiento lo hacen formando parte de un planteamiento multimodal30,31.
La mayoría de los opioides se metabolizan en el hígado a través de la glucuronidación o la oxidación vía citocromo P450. Estos procesos están alterados en los pacientes con CH, lo que condiciona una disminución de la depuración y un aumento de la BD, produciéndose aumento de la vida media y acumulación del fármaco4. Curiosamente, la alteración de su metabolismo en las diversas isoenzimas del sistema citocromo P450 puede ocasionar que disminuya su efecto analgésico9,32.
En la enfermedad hepática y particularmente en la colestasis varios opioides endógenos se sobreexpresan. Este incremento del «tono opioidoérgico» está asociado con el prurito, la encefalopatía, la vasodilatación, la disminución del flujo sanguíneo esplácnico, y la retención de agua y sodio presentes en los pacientes con enfermedad hepática avanzada8. El aumento de la expresión de opioides endógenos hace a estos pacientes más lábiles a la administración de opioides exógenos.
La indicación de fármacos opiáceos debe ser restringida y bajo una monitorización rigurosa por la alta incidencia de efectos secundarios graves33. Es necesario monitorizar signos como la sedación y/o estreñimiento que predisponen a la aparición de la EH, situación que se presenta con más frecuencia en personas con encefalopatía previa o HTP6,37. En los pacientes alcohólicos el riesgo de adicción está aumentado4.
Como regla general, los opioides más lipofílicos como el fentanilo y la metadona son más impredecibles en su BD así como los que tienen un gran metabolismo de primer paso como la morfina, ya que son más sensibles al cortocircuito portosistémico33. Los opioides que requieren metabolismo oxidativo, como la oxicodona y la meperidina, presentan depuración afectada en fases tempranas de la enfermedad hepática33.
MorfinaEs el agonista μ con el cual se comparan el resto de opiáceos. Su BD es muy variable, de 35-75%, e inicio de acción lenta. Su efecto analgésico es más largo que su vida media por su baja solubilidad y lenta eliminación cerebral31. Es metabolizado en el hígado por glucuronidación en morfina-3-glucorónido (M3G) y morfina-6-glucorónido (M6G), responsables de su toxicidad35. En presencia de IR, altas dosis o tratamiento crónico existe riesgo de acumulación de sus metabolitos, por lo que se debe evitar en pacientes con SHR o IR8. La presencia de cortocircuito portosistémico condiciona una alta BD de la morfina oral que se puede equiparar a la BD de la morfina intravenosa33. En la CH la vida media de este fármaco está aumentada aproximadamente el doble comparado con controles sanos34. El M6G es el principal responsable de la intoxicación produciendo alteraciones en SNC, depresión respiratoria y mioclonías35, por lo que las dosis se deben espaciar y/o disminuir.
HidromorfonaPotente agonista μ, análogo de la morfina. Su potencia es de 5 veces mayor que la morfina cuando se administra por vía oral y 7 veces más administrado por vía parenteral. Es muy hidrofílica, con menos incidencia de prurito, sedación, náuseas y vómitos que la morfina. Tiene metabolización hepática en hidromorfona-3-glucurónido (H3G), metabolito que no tiene actividad analgésica pero sí actividad neuroexcitatoria, aunque se produce en pequeñas cantidades y solo se acumula si hay IR35. Por disminución del metabolismo de primer paso tiene mayor BD y se ha de iniciar en bajas dosis y lentamente8,36. Al no producir metabolitos tóxicos y ser mejor tolerada que otros opioides está indicada en el paciente con enfermedad hepática sin SHR36.
FentaniloEs agonista μ opioide muy lipofílico, con rápido inicio de acción y más potente que la morfina. Su lipofilidad permite la aplicación transdérmica a través de parches para el dolor continuo o transmucoso para el dolor irruptivo31. Tiene alta unión a proteínas plasmáticas, por lo que se necesita menos dosis6,8. Libera menos histamina, lo que condiciona mejor estabilidad hemodinámica. No tiene metabolitos tóxicos. Es de elección en el paciente con CH4,6,8.
Parche de fentaniloÚtil en pacientes que no toleran la vía oral, tienen alteraciones para la absorción gastrointestinal o no tienen adherencia a la medicación oral. La dosificación puede ser dificultosa por las variaciones interindividuales de la absorción transdérmica por sudoración, tejido adiposo o la temperatura de la piel31. Los pacientes con CH presentan alteración de la permeabilidad de la piel y el flujo sanguíneo cutáneo lo que puede favorecer a una absorción errática del fármaco8.
Citrato de fentanilo transmucosoRápido inicio de acción (5-10 min) y corta duración; la vía transmucosa evita el metabolismo de primer paso. De elección en el dolor irruptivo31.
MetadonaEs una opción interesante en el tratamiento del dolor crónico por su bajo coste, alta BD, afinidad a múltiples receptores, lenta eliminación (vida media 26.8h) sin metabolitos activos31. Tiene poca afinidad por los receptores μ cuando se compara con la morfina, lo que explica la menor incidencia de los efectos secundarios relacionados con este receptor31. Al ser antagonista de los receptores NMDA e inhibir la recaptación de noradrenalina puede ser útil en el tratamiento del DN31. Tiene alta unión a proteínas plasmáticas, por lo que se precisan dosis menores4. Es muy lipofílica, lo que explica su extensa distribución en los tejidos31. La ausencia de metabolitos neurotóxicos hace que sea considerada como fármaco de segunda línea en pacientes con predisposición a presentar alteraciones por neurotoxicidad4. Hay estudios que señalan que es segura en la disfunción hepática6,9. Como desventaja tiene una farmacocinética y farmacodinámica impredecible con gran variabilidad interindividual6,31. Es utilizada como fármaco de mantenimiento en pacientes con adicción crónica a los opioides, población que tiene una gran incidencia de VHC y por ende de CH; en esta población no se han evidenciado alteraciones en la disposición del fármaco6,8. No se acumula en presencia de insuficiencia renal y no se filtra en hemodiálisis6,8,31.
BuprenorfinaEs un agonista parcial μ opioide. Por su alto metabolismo de primer paso no se administra por vía oral. Su eliminación es principalmente fecal, por lo que puede ser útil en pacientes con SHR7, aunque se puede acumular en presencia de colestasis4. Se han descrito casos de toxicidad hepática por buprenorfina, más frecuente tras el uso intravenoso6. Existen resultados contradictorios respecto a la hepatotoxicidad en pacientes hepatópatas, particularmente en hepatitis C, con estudios que demuestran aumento de las transaminasas y otros estudios que no confirman este hecho6,8. Durante el tratamiento, se recomienda monitorizar los niveles de enzimas hepáticas6.
Opioides a evitar en pacientes con cirrosis hepáticaOxicodonaEs predominantemente un profármaco convirtiéndose en oximorfona, a través de la metabolización hepática en el sistema citocromo P45033. Presenta una BD del 60%, vida media corta (2,5-4h), fácil dosificación y rápido mecanismo de acción31. Tiene una presentación de liberación retardada de 2 dosis/24h. Posee actividad intrínseca analgésica a través de los receptores kappa por lo que se ha descrito su utilidad en el tratamiento del DN31. Es uno de los opioides más prescritos y de los cuales hay más mal uso o abuso31.
MeperidinaEs metabolizada por el citocromo P450 en normeperidina, metabolito tóxico con efectos neuroexcitatorios y convulsionantes, que se acumula en presencia de IR4,31.
Codeína, hidrocodonaSon profármacos cuyo efecto analgésico depende de la conversión hepática por el citocromo P450 en morfina e hidromorfona, respectivamente4,6,8,31,33.
TramadolEs una opción válida en el tratamiento del dolor moderado a severo del paciente con CH4. Actúa como agonista no selectivo de los receptores μ opioides e inhibiendo la recaptación de noradrenalina y serotonina15,37. Está disponible en formas de liberación inmediata y retardada, tiene techo analgésico y sus efectos adversos más comunes son estreñimiento, náuseas, vómitos, somnolencia y prurito31. Su combinación con paracetamol es efectiva en el tratamiento del dolor agudo38. La dosis diaria no debe exceder los 400mg y disminuir en presencia de IR4,15. Puede provocar dependencia psíquica y síndrome de abstinencia15. Se ha asociado con convulsiones cuando se exceden las dosis recomendadas o se combina con inhibidores selectivos de la recaptación de serotonina, inhibidores de la monoaminoxidasa o neurolépticos, y se ha vinculado con el síndrome serotoninérgico4,15. Puede producir estreñimiento por su agonismo μ y sus efectos anticolinérgicos, lo que puede predisponer a la EH. La dosis recomendada en el paciente con CH es de 25mg/8h6.
TapentadolEs un nuevo analgésico que actúa como agonista μ opioide e inhibidor de recaptación de noradrenalina, por lo que es útil en el tratamiento del DN39. Tiene un extenso metabolismo de primer paso, con una BD oral en ayuno de 32%. Se metaboliza en el hígado, principalmente por conjugación, y tiene escasa unión a proteínas plasmáticas por lo que presenta bajo potencial de interacciones farmacológicas, no produce metabolitos activos y tiene eliminación renal39. Es eficaz en el dolor lumbar crónico, osteoartrosis de cadera o rodilla y PND40. Comparado con agonistas opioides μ tiene menor incidencia de náuseas, vómitos, estreñimiento, prurito y dependencia física; no se ha evidenciado depresión respiratoria y tiene mejor adherencia al tratamiento39,40. Parece ser una buena opción en el tratamiento del dolor en pacientes con enfermedad hepática avanzada, a pesar del número limitado de estudios. En estos pacientes, probablemente la BD del fármaco está aumentada, por lo que se recomienda comenzar con la menor dosis (25-50mg) y alargar el intervalo cada 24h6.
AnticonvulsionantesSu uso deriva del hecho que la hiperexcitabilidad neuronal es una de las características de la epilepsia y del DN. Los anticonvulsionantes actúan modulando la neurotransmisión periférica y central a través de varios mecanismos de acción como el aumento de la actividad GABAérgica, disminución de la actividad glutaminérgica, bloqueo de los canales de sodio y calcio dependientes del voltaje o la alteración de las vías intracelulares41. Múltiples estudios señalan a los canales de sodio y calcio como el eslabón fundamental para la producción y mantenimiento del DN, principalmente la subunidad α2δ de los canales de calcio dependientes del voltaje41,42. La eficacia de los anticonvulsionantes es distinta según el tipo del DN; la mayoría de los estudios están realizados en polineuropatía periférica (PNP) y neuralgia del trigémino. Hay pocos estudios en DN asociado a una radiculopatía, probablemente el DN más prevalente42. Muchos anticonvulsionantes son metabolizados por el sistema del citocromo P450 y excretados por vía renal, por lo que en los pacientes con enfermedad hepática avanzada pueden ser efectivos en dosis menores y menos frecuentes4. La fenitoína, la carbamacepina y el ácido valproico son hepatotóxicos y están contraindicados en la CH6,7.
Bloqueantes de la subunidad α2δ de los canales de calcio dependientes de voltajeForman parte de la primera línea de tratamiento del DN16. No poseen metabolismo hepático y tienen escasa unión a las proteínas plasmáticas6. Se pueden utilizar en caso de CH, aunque se ha comunicado hepatotoxicidad. Se deben titular lentamente, a dosis bajas para evitar la sedación y mareo, efectos secundarios más frecuentes5. Por su eliminación renal se deben de vigilar en caso de SHR6,17
GabapentinaEs el fármaco de elección en el inicio del tratamiento del DN a dosis de 300-1.200mg/8h. Efectivo en varios tipos de DN, a partir de 1.200mg/día43. Se ha comunicado hepatotoxicidad44.
PregabalinaCon una farmacocinética lineal se utiliza cada 12 h (150-600mg/día). La dosis de mayor efectividad son 300-600mg/día y las enfermedades que mejor responden son la NPH y la NPD45. En la radiculopatía lumbosacra tiene un efecto moderado de mejoría46. Se han reportado casos de fallo hepático agudo idiosincrásico47,48. A pesar de la similitud farmacológica, pacientes que no responden a la gabapentina pueden responder a la pregabalina49.
Topiramato y lamotriginaTienen metabolismo hepático, por lo que se debe disminuir la dosis6,7,50. El topiramato actúa bloqueando los canales de sodio, GABA agonista y antagonista de los receptores AMPA, por lo que aumenta el glutamato. Se ha señalado su utilidad en la neuralgia del trigémino y en el dolor por compresión radicular. De los efectos adversos destaca la litiasis renal y el glaucoma (inhibidor de la anhidrasa carbónica)51. La lamotrigina bloquea los canales de sodio e inhibe la liberación de glutamato. Están indicados en la neuralgia del trigémino, dolor por frío, PND y en la polineuropatía asociada a VIH. Pueden ser alternativa a otros tratamientos52.
Por último, los nuevos anticonvulsionantes (lancosamida, levetiracetán) no han mostrado efectividad en el tratamiento del DN, sin embargo a pesar de la falta de respuesta en ensayos clínicos controlados, algunos subgrupos de pacientes pueden responder a estos fármacos.
Anticonvulsionantes a evitar en pacientes con cirrosis hepáticaCarbamacepina y oxcarbamacepinaActúan bloqueando los canales de sodio dependientes del voltaje. Muy utilizadas en la neuralgia del trigémino, sin suficiente evidencia que apoye su uso como fármacos de primera línea53. Tienen múltiples efectos secundarios destacando mareos, sedación, anemia aplásica, agranulocitosis, ataxia e hiponatremia. Son hepatotóxicas, ya que son grandes inductoras de enzimas hepáticas ocasionando deterioro de la función hepática en pacientes con CH4,6,17,50,51,54.
BenzodiacepinasAunque su toxicidad hepática solo se ha reportado de manera idiosincrásica, su uso en pacientes hepatópatas está limitado por su vinculación con la aparición de la EH54. Tienen poca evidencia que justifique su uso en el DN42.
AntidepresivosLos antidepresivos tricíclicos (ADT) y los inhibidores selectivos de la recaptación de serotonina y norepinefrina (ISRSN) forman parte de la primera línea en el tratamiento del DN16. Su efecto analgésico está mediado por la inhibición de la recaptación de noradrenalina, ya que este neurotransmisor en la hendidura sináptica fortalece los mecanismos de supresión del dolor en la vía descendente del dolor55,56. Tienen metabolismo hepático a través del citocromo P450, múltiples interacciones medicamentosas y eliminación renal. Se han de dosificar lentamente para disminuir los efectos secundarios, que pueden aparecer antes que el efecto analgésico55,56. Además de las propiedades analgésicas de los antidepresivos, debemos recordar que la depresión tiene una alta incidencia en los pacientes que sufren de dolor crónico55,56.
Antidepresivos tricíclicosSon los más eficaces en el tratamiento del DN, aunque el 20% de los pacientes abandona el tratamiento por los efectos secundarios16,56. Los efectos adversos están relacionados con su acción antihistamínica y anticolinérgica, siendo los más frecuentes: sedación, hipotensión arterial ortostática, aumento de peso, boca seca, visión borrosa, retención urinaria, alteraciones en la esfera sexual, sudoración, confusión, delirio, prolongación del intervalo QT, aumento de riesgo de muerte súbita y síndrome de abstinencia55,56. Los efectos anticolinérgicos más frecuentes en los pacientes con CH con mayor riesgo de EH4,6. La amitriptilina (10 a 25mg/24h) e imipramina son los más sedantes, mientras que la desipramina y nortripitilina tienen menos efectos secundarios6,57.
Inhibidores selectivos recaptación de noradrenalina y serotoninaTienen buena absorción oral y gran BD. Aunque se ha descrito hepatotoxicidad se deben considerar en pacientes con enfermedad cardíaca como alternativa a los ADTC55,57. Como efectos adversos destacan náuseas, vómitos, disfunción sexual, boca seca, sudoración, estreñimiento, hipotensión ortostática, síndrome de secreción inadecuada de hormona antidiurética. Se ha descrito síndrome serotoninérgico56. En pacientes con enfermedad hepática crónica el metabolismo de la venlafaxina está disminuido un 50%, y en la enfermedad avanzada puede llegar a un 90%6. La dosis de inicio es 37,5mg/12h. En dosis altas puede producir HTA y arritmias cardíacas57. La duloxetina es hepatotóxica, por lo cual su uso se debe evitar6,57.
Tratamiento farmacológico tópicoPor su actividad localizada y su baja absorción sistémica tienen un perfil de seguridad favorable y bajo riesgo de interacciones farmacológicas. Existe evidencia científica que respalda la eficacia y seguridad de estos agentes en muchos síndromes dolorosos incluidos el DN58. Los fármacos más utilizados son los anestésicos locales y la capsaicina.
Anestésicos localesActúan a través del bloqueo de los canales de sodio disminuyendo la actividad de las neuronas primarias aferentes lesionadas, lo que atenúa la sensibilización central, disminuyendo el dolor espontáneo y evocado con atenuación de la alodinia e hiperalgesia59.
EMLA (euthetic mixture of local anesthetics). Es una crema que mezcla lidocaína al 2,5% con prilocaína al 2,5%. Es efectiva en la NPH58.
Parche de lidocaína al 5%. Es eficaz en la NPH y la PND, forma parte del primer escalón del tratamiento del DN15. Proporciona una barrera contra la estimulación mecánica que provoca la alodinia59. Las concentraciones plasmáticas son mínimas por lo que hay un exiguo riesgo de absorción sistémica59.
CapsaicinaEs un agonista exógeno del receptor potencial transitorio vaniloide 1 (TRPV1), receptor que se expresa en los nociceptores que responden a la temperatura, cambios de pH o lípidos endógenos. La activación de estos receptores por un agonista químico estable como la capsaicina puede generar daño de la función nociceptora local producido por disfunción de los mismos o por la degeneración de las terminales nerviosas. Actúa en la piel y mejora el dolor disminuyendo la actividad espontánea y la hipersensibilización, y no está influido por la absorción sistémica del fármaco60. Inicialmente produce aumento de la sensibilidad y del dolor, disminuyendo progresivamente por reducción en la expresión de los TRPV1. Se presenta en cremas o lociones de bajas concentraciones, 0,025 a 1%, y parches tópicos al 8%. Los principales efectos adversos están relacionados con la irritación local transitoria60. No necesita disminución de la dosis en pacientes hepatópatas60.
Técnicas intervencionistas para el tratamiento del dolorSon una opción segura, eficaz y/o complementaria en el tratamiento multidisciplinario del dolor. Pueden aliviar el dolor por largos períodos de tiempo y son útiles en enfermedades de difícil diagnóstico, o para decidir y/o planificar una intervención quirúrgica determinada. El hecho de que permitan reducir el consumo de analgésicos, disminuyendo las interacciones farmacológicas y la aparición de efectos adversos, posicionan a esta técnicas como una valiosa herramienta para el manejo del dolor del paciente con CH.
El cuestionamiento de la conveniencia de realizar procedimientos terapéuticos invasivos en pacientes con alteraciones de la coagulación está actualmente controvertido. Aunque es bien sabido que en los pacientes con CH las pruebas de laboratorio habituales como INR y aPTT no dan información sobre riesgo de sangrado, determinadas técnicas como la implantación de catéteres neuroaxiales no son aconsejables. Los bloqueos periféricos continuos así como la administración neuroaxial de dosis única de un fármaco de larga duración son técnicas seguras y no plantean la necesidad de corrección de la coagulación.
Describimos de manera breve las técnicas intervencionistas más utilizadas61,62:
- 1.
Bloqueos nerviosos diagnósticos y terapéuticos. Consisten en administrar anestésicos locales en la periferia de los nervios implicados en el dolor para bloquear la conducción nerviosa y evitar la transmisión del dolor al SNC. Frecuentemente son un paso previo para procedimientos más definitivos. Dentro de este grupo destacamos los bloqueos para el diagnóstico y tratamiento del dolor axial de origen facetario.
- 2.
Bloqueo epidural. Útil en el diagnóstico y tratamiento del dolor ocasionado por la irritación de una raíz nerviosa, se basa en depositar el fármaco (habitualmente corticoides) en el espacio epidural lo más cerca posible de las estructuras afectadas. Se ha utilizado con cierto beneficio en síndromes dolorosos, como la cirugía fallida de espalda y/o la estenosis de canal.
- 3.
Infiltración de puntos gatillo. Utilizada en el dolor miofascial, se basa en la infiltración del punto cuya palpación desencadena el dolor (puntos gatillo). Bloquea el estímulo doloroso y produce relajación muscular.
- 4.
Infiltración de articulaciones dolorosas. Se administran fármacos que buscan disminuir la inflamación, aliviar el dolor y ayudar a la fisioterapia. Las articulaciones más intervenidas son hombro, cadera, rodilla y articulación sacroilíaca.
- 5.
Radiofrecuencia. Consiste en hacer pasar una corriente eléctrica de baja energía y alta frecuencia a través de 2 electrodos, el electrodo activo que se encuentra en el extremo de una aguja que se utilizara para abordar el área que se desea tratar, y el electrodo dispersivo, placa de material conductivo. El paso de corriente a través de este circuito genera calor en la punta del electrodo activo y realiza el tratamiento. Las indicaciones más frecuentes son el síndrome facetario y sobre distintos nervios periféricos.
- 6.
Bloqueos del sistema nervioso simpático. El sistema simpático está involucrado en el origen y mantenimiento de varios síndromes dolorosos (síndrome regional complejo, DN, miembro fantasma, etc.), en enfermedades relacionadas con una disminución del flujo sanguíneo (angina isquémica, enfermedad vascular periférica) y en el dolor visceral incontrolable (cáncer de páncreas). Los bloqueos del sistema nervioso simpático son una opción diagnóstica terapéutica posible y eficaz. En algunos casos, para el control prolongado del dolor se produce lesión del nervio afectado, mediante fármacos o temperatura. Los bloqueos simpáticos más comunes son: ganglio estrellado, simpático torácico y lumbar, plexos celíaco e hipogástrico y ganglio impar.
- 7.
Dispositivos implantables. Se deben reservar para el tratamiento del dolor de fuerte intensidad resistente a múltiples tratamientos. Aunque sin evidencia científica demostrada, es probable que los pacientes con enfermedad hepática avanzada puedan tener más riesgo de complicaciones asociadas a estos dispositivos.
- -
Neuroestimuladores. Se basan en la acción de un dispositivo que emite señales eléctricas a un electrodo colocado en la zona de los nervios que transmiten el dolor, bloqueando su conducción. Su principal indicación es la estimulación de los cordones posteriores medulares para tratar el síndrome de cirugía de espalda fallida, síndrome regional complejo, miembro fantasma, etc. Recientemente se utilizan con buenos resultados, la neuroestimulación de nervios periféricos y la neuroestimulación subcutánea.
- -
Analgesia neuroaxial continua. La administración de pequeñas dosis de fármacos lo más cerca posible de los receptores permite una analgesia potente con escasos efectos secundarios sistémicos. Los pacientes con una expectativa de vida superior a 3 meses son candidatos a tratamiento intratecal mediante un catéter intraespinal y sistema de infusión subcutánea o externa si la expectativa de vida es menor. Los fármacos más utilizados son la morfina, hidromorfona, bupivacaína y clonidina.
- -
- 1.
Neuroestimulación eléctrica transcutánea (TENS). Consiste en administrar una corriente eléctrica a través de electrodos colocados en la piel del área afectada, lo que estimula selectivamente fibras nerviosas periféricas bloqueando la transmisión del dolor. Es fácil de aplicar con pocos efectos secundarios62.
- 2.
Iontoforesis. Se fundamenta en la utilización de una corriente directa para mejorar la disposición del fármaco en la piel, mediante la aplicación de un electrodo a sustancias ionizables como los anestésicos locales y/o corticoides63.
- 3.
Acupuntura. Se basa en la inserción y estimulación de agujas en distintos puntos del cuerpo. Aunque inicialmente solo era parte de la medicina tradicional china, en la actualidad muchos practicantes entienden este tratamiento en términos fisiológicos sin referencia a los conceptos antiguos64. Existen varios estudios que muestran beneficio en el dolor crónico y se ha utilizado con seguridad en pacientes con enfermedad hepática65.
- 4.
Intervenciones psicológicas. Existe evidencia que soporta el uso de la terapia cognitiva-conductual, biofeedback, técnicas de relajación, terapia de grupo y psicoterapia en el dolor crónico. Estas técnicas deben ser consideradas como parte del tratamiento multimodal18.
- 5.
Rehabilitación y terapia física. De igual manera, es efectiva para el tratamiento del dolor crónico y permite recuperar o mejorar las limitaciones ocasionadas por el dolor. Deben estar presentes como parte del tratamiento multimodal18.
- •
No hay guías basadas en la evidencia para el tratamiento del dolor en pacientes con CH.
- •
El enfoque del tratamiento del dolor es distinto en función de si es es agudo o crónico. El éxito del tratamiento está directamente relacionado con el conocimiento de la fisiopatología del dolor. Se debe individualizar el tratamiento a cada paciente.
- •
El paracetamol es seguro, se recomiendan dosis < 3 g día máximo.
- •
Los AINE se deben contraindicar por el riesgo de insuficiencia renal aguda y/o sangrado gastrointestinal
- •
La prescripción de opioides se ha de evitar en pacientes con riesgo de encefalopatía. Su indicación condiciona un seguimiento exhaustivo de los pacientes.
- •
Los opioides con mejor perfil farmacológico son el fentanilo y la hidromorfona, a dosis más bajas y menos frecuentes. Se puede considerar en ciertos pacientes la metadona.
- •
El tramadol es una opción segura. El tapentadol es una buena opción terapéutica ya que tiene pocas interacciones farmacológicas y menos efectos gastrointestinales que otros agonistas μ opioides.
- •
Para el tratamiento del DN, el medicamento oral de elección es la gabapentina. Se puede considerar el uso de antidepresivos tricíclicos como la amitriptilina.
- •
El tratamiento tópico es de gran utilidad ya que puede disminuir el consumo de fármacos analgésicos sistémicos, disminuyendo las interacciones farmacológicas y la posibilidad de presentar efectos adversos asociados.
- •
Las técnicas intervencionistas son de gran interés en el tratamiento del dolor moderado a severo ya que permiten disminuir el tratamiento farmacológico oral y con ello los efectos adversos.
- •
Considerar las intervenciones psicológicas, la terapia física y la rehabilitación como parte de la terapia multimodal en el abordaje del dolor crónico.
Los autores declaran no tener ningún conflicto de intereses.
Los autores agradecen a la Dra. Pilar Taurá Reverter su inestimable colaboración en la realización de esta revisión.

