





Casos Clínicos en Gastroenterología y Hepatología
Más datosEl síndrome de Currarino es una enfermedad hereditaria autosómica dominante asociada a mutaciones en el gen HLXB9, caracterizada por la asociación de malformaciones óseas sacrococcígeas, tumores presacros y malformaciones anorrectales, cuya presentación de forma completa es muy rara1,2. Fue descrita por primera vez en 1981 en una serie de casos diagnosticados en la infancia que presentaban tumores presacros como teratomas, meningoceles o quistes entéricos3. Hasta la fecha se han publicado muy pocos casos en los que se asocie la presencia de tumores neuroendocrinos a este síndrome4.
Presentamos el caso de una mujer de 27 años con antecedentes de agenesia sacrococcígea parcial e intervenida en la infancia por imperforación anal.
Acude por el hallazgo de una masa retrorrectal en una resonancia magnética (RM) pélvica realizada en el contexto de un estudio por estreñimiento pertinaz.
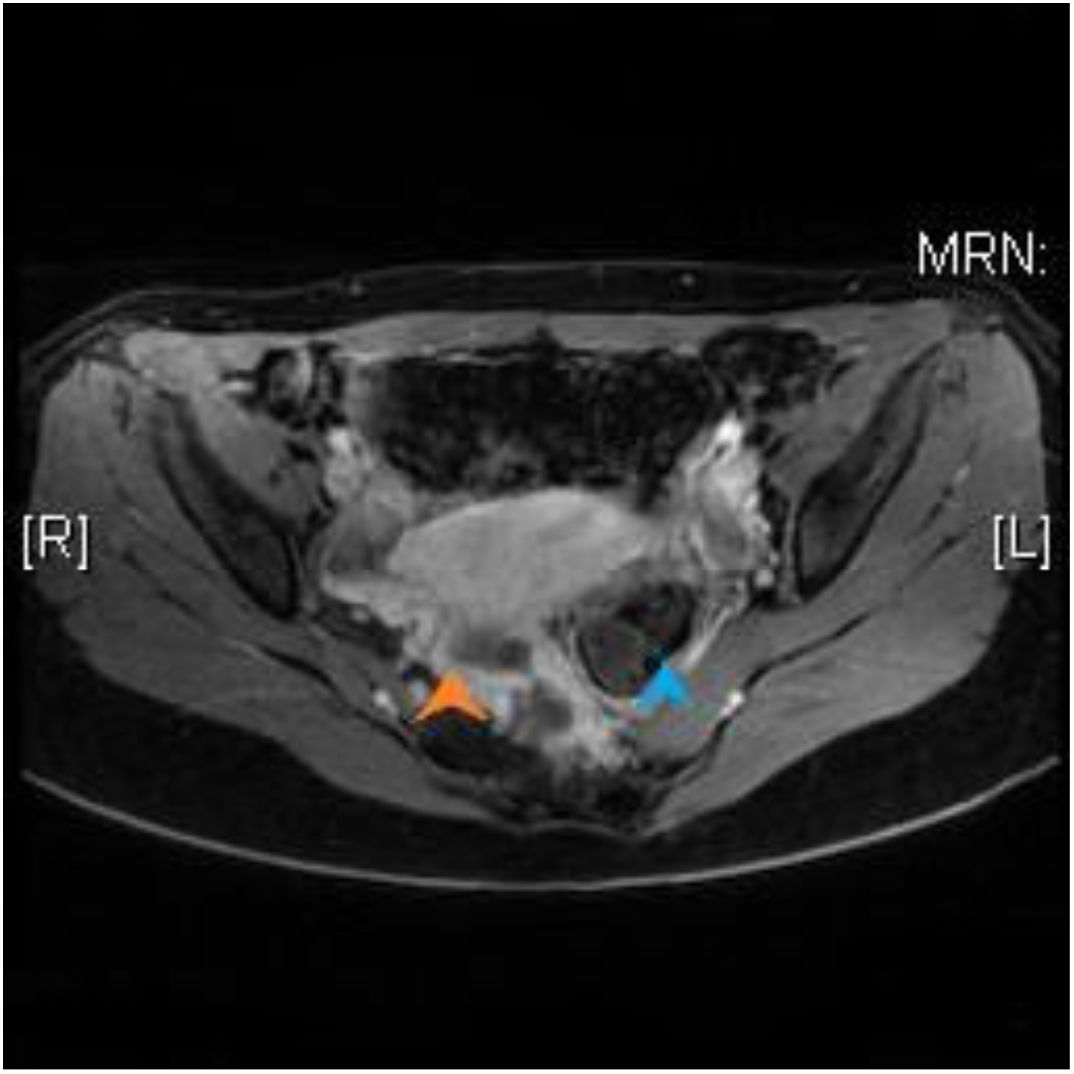
En la RM se objetiva una lesión heterogénea adyacente a la pared lateral derecha del recto en el espacio supraelevador, constituida por lesiones quísticas y sólidas, compatible con un hamartoma. La lesión desplaza el recto sin signos de invasión del mismo (fig. 1).
 con aspecto heterogéneo y la presencia de lesiones quísticas, que lateraliza el recto (flecha azul) hacia la izquierda.")
Se observa también una agenesia del coxis e hipoplasia con desplazamiento hacia el lado izquierdo de la última vértebra sacra, con atrofia de los músculos piramidal y elevador del ano derechos.
Realizamos la extirpación de la lesión mediante un abordaje retrorrectal transacro según la técnica de Kraske, sin incidencias ni necesidad de resecar el coxis o las últimas vértebras sacras debido a la hemiagenesia que presenta esta paciente, logrando extirpar la lesión. El postoperatorio se desarrolla sin complicaciones y la paciente es dada de alta a los 4 días de la intervención.
El análisis anatomopatológico revela que se trata de un tumor neuroendocrino grado 1 (NET-G1/carcinoide) asociado a quiste tailgut (hamartoma quístico retrorrectal). En el estudio inmunohistoquímico la celularidad neoplásica muestra diferenciación neuroendocrina con positividad para citoqueratina AE1/AE3, sinaptofisina, CD56 y focalmente para cromogranina. El índice de proliferación tumoral (Ki-67) es<1%.
El síndrome de Currarino es una enfermedad muy infrecuente que se suele diagnosticar en la infancia; se caracteriza por la tríada de malformación anorrectal, masa presacra y defecto óseo sacrococcígeo.
La clínica es variada, aunque en la mayoría de los casos se asocia a meningitis, infecciones urinarias recurrentes, incontinencia urinaria y estreñimiento crónico1; no obstante, es frecuente que algunos pacientes permanezcan asintomáticos.
El diagnóstico es radiológico mediante RM y tomografía computarizada para determinar las lesiones óseas y la presencia de la masa presacra.
Las lesiones sacrococcígeas más frecuentes son la hemiagenesia o lateralización de los cuerpos vertebrales sacros y la agenesia coccígea. Uno de los hallazgos radiológicos patognomónicos es el «signo de la cimitarra», que se observa en las proyecciones anteroposteriores de una radiografía simple de pelvis.
Las malformaciones anorrectales tienen una gran variabilidad, con casos de imperforación anal como el que presentamos, duplicaciones rectales, o fístulas enteroespinales, rectovaginales o rectovesicales1.
En cuanto a la masa presacra que asocian los pacientes con síndrome de Currarino, se han descrito un 60% de meningoceles, un 25% de teratomas y un 15% de otros tumores como hamartomas, lipomas o quistes epidérmicos1,4.
La presencia de tumores neuroendocrinos presacros es extremadamente rara, con únicamente unos pocos casos publicados asociados al síndrome de Currarino4. Dada la baja incidencia de este tipo de tumores es difícil determinar cuál es su evolución y pronóstico.
Ante la baja prevalencia de malignidad en los tumores de esta localización, no se consideró la opción de realizar una biopsia preoperatoria, ya que el tratamiento se estableció como quirúrgico desde un primer momento y la realización de una biopsia preoperatoria conlleva un elevado riesgo de diseminación tumoral2.
El tratamiento de elección tanto de los hamartomas como de los tumores neuroendocrinos es la extirpación quirúrgica, prefiriendo el abordaje de Kraske por su menor agresividad y recuperación más rápida.
Al tratarse de una enfermedad hereditaria autosómica dominante esta paciente debería realizar un adecuado asesoramiento genético, que está pendiente de resultados en otro centro, teniendo en cuenta que el riesgo de transmitir la enfermedad es del 50% y que se trata de una mujer joven en edad fértil5.
En el caso que presentamos es de vital importancia continuar con un seguimiento estrecho mediante pruebas de imagen como la RM por el riesgo de recidiva del tumor neuroendocrino.

