




Alagille syndrome (AS) (ALGS; OMIM 118450) has an approximate incidence of 1/30000 live births.1 It is a multisystemic disease with an autosomal dominant pattern of inheritance and almost complete penetrance, although around 50–70% of cases are sporadic and caused by de novo mutations; furthermore, clinical expression varies greatly. It is a syndrome characterised by hepatic manifestations, mainly cholestasis (96%), butterfly vertebrae (51%), posterior embryotoxon (78%) and congenital heart disease, the most common of which is pulmonary artery stenosis (97%), and peculiar facies (96%). Histopathology study of the liver biopsy shows a paucity of interlobular bile ducts. The defect has been localised in 2 different genes. Mutations in the Jagged1 (JAG1) gene have been identified in more than 90% of cases.1–3 Prognosis essentially depends on the liver impairment and cardiovascular malformations.4,5 There is usually some spontaneous improvement in the early years of life, although 15–20% of patients will require liver transplantation.5,6 This report describes a clinical case of special interest due to the association of AS with intestinal atresia in the neonatal period, reported in very few cases in the literature, and the diagnosis of the child's father as a result of his son's diagnosis, with much less severe expression.
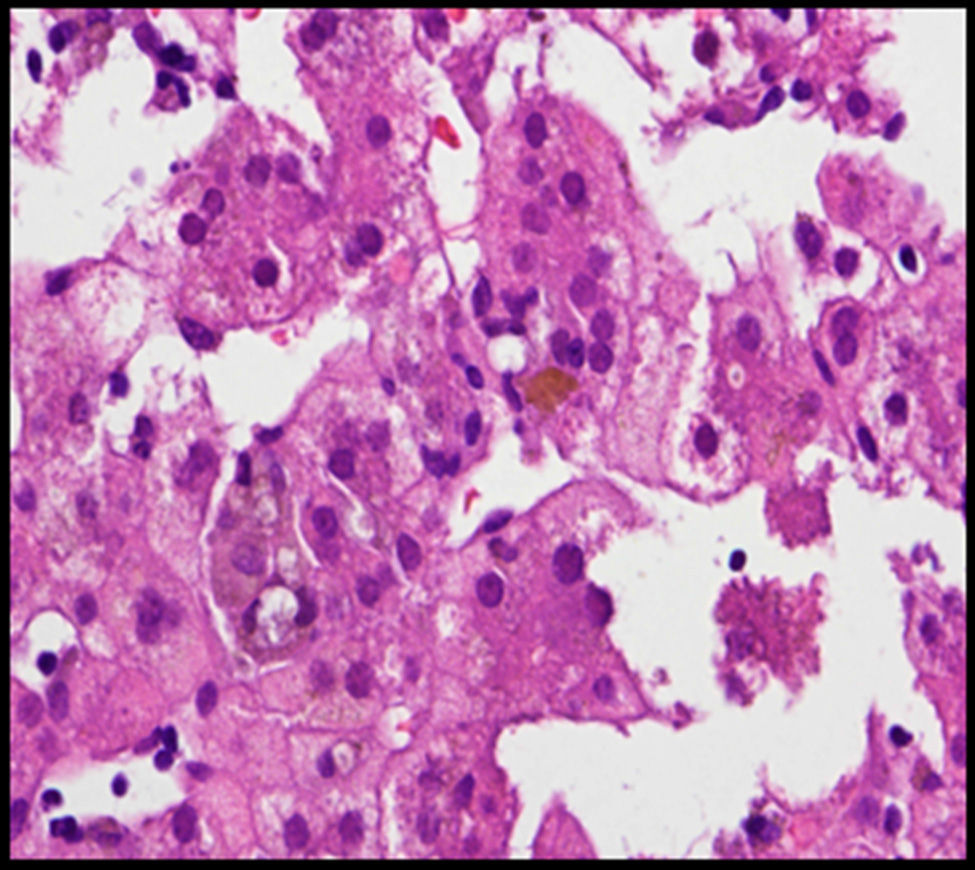
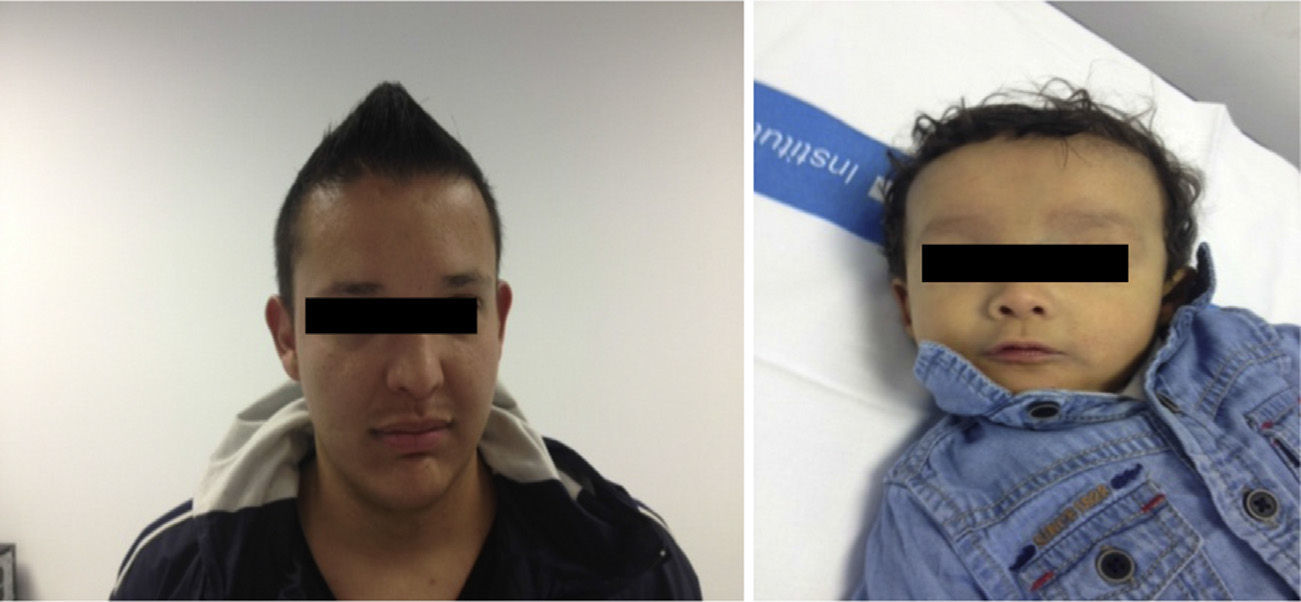
The case involved a full-term newborn, of adequate weight, with pulmonary branch stenosis detected due to heart murmur. Difficulty feeding, vomiting, and abdominal distension with failure to pass the first meconium were obvious from the first hours of life. Suspecting intestinal atresia, abdominal X-ray was performed, revealing absence of rectal gas and distended loops in the upper portion of the abdomen, confirming the diagnosis. Surgery was performed 48h after birth, with resection of 23cm of small bowel proximal to the area of stenosis. The infant received parenteral nutrition for 11 days. From day 3, he presented non-isoimmune jaundice with maximum total bilirubin of 12.5mg/dL. At day 10, a cholestatic pattern of liver enzymes was observed (total bilirubin 6.41mg/dL, direct bilirubin 4.75mg/dL, gamma-glutamyl transferase [GGT] 290U/L, aspartate aminotransferase [AST] 48U/L, alanine aminotransferase [ALT] 26U/L), attributed to parenteral nutrition, which was discontinued as a result. After persistence of the cholestasis pattern in follow-up laboratory tests, abdominal ultrasound was performed, in which a full gallbladder was observed, with no bile duct dilatation. Gradual worsening of the cholestasis was noted, with maximum direct bilirubin levels increasing to 10mg/dL, so the aetiological study was extended. Metabolic study, hormone levels, sweat test, cytomegalovirus testing and alpha-1 antitrypsin were all normal. Magnetic resonance cholangiography (MRC) showed no extrahepatic obstructive signs or obstruction of the main hepatic branches. Hepatobiliar iminodacetic acid (HIDA) scan showed passage of contrast to the duodenum. Liver biopsy found paucity of bile ducts (Fig. 1) which, associated with the cholestasis, heart disease and peculiar facies (Fig. 2), suggested a diagnosis of AS. Ophthalmological and vertebral X-ray were normal. The cholestasis pattern gradually worsened (maximum GGT levels of 1500U/L) with conjugated hyperbilirubinaemia around 12mg/dL despite treatment with ursodeoxycholic acid, and severe hypercholesterolaemia (maximum 450mg/dL), with appearance of difficult-to-control pruritus, together with progressive splenomegaly secondary to portal hypertension. The child required liver transplant from a live parent (mother) at 2 years of age, which was uneventful, with good progress. Analysis of the JAG1 gene detected heterozygous C-to-A transversion (c.756>A) which presumably, at protein level, results in a premature stop codon (p.Tyr255*), described previously in the literature as a mutation associated with AS.7,8
.")

The father (27 years old) had peculiar facies (Fig. 2) and a history of left pulmonary branch stenosis that required surgery in childhood, with no haemodynamic repercussion at present, and anicteric cholestasis (AST/ALT 130/165U/L, GGT 968U/L, alkaline phosphatase 442U/L, bilirubin 0.83mg/dL, cholesterol 210mg/dL) detected at age 21 as an incidental finding on a routine blood test and attributed to alcohol abuse. As a result of his son's diagnosis, MRC was performed, which found a small-calibre extrahepatic bile duct with absence of visualisation of the intrahepatic bile duct. Liver biopsy showed absence of bile ducts in more than 50% of the portal spaces, bile duct hypoplasia and intrahepatocytic haemosiderosis. Due to the diagnosis of AS in the child, the father was also diagnosed, although he presented less severe expression. The variability in the clinical expression within the family should be highlighted, with major liver impairment in the child and, in contrast, very mild impairment in the father at the age of 27 (Figs. 1 and 2).
The association with intestinal atresia is a manifestation that does not appear in most descriptions of this syndrome. After an exhaustive literature review, we found very few cases that associate AS with intestinal malformations.9 The JAG1 gene is one of the components of the NOTCH signalling pathway, which plays a key role in angiogenesis. The intestinal atresia associated with AS could be the result of an alteration in vascularisation during the embryonic development of the digestive tract.10
Please cite this article as: González Pastor S, Montraveta Querol M, del Alcazar Muñoz R, Ojanguren Sabán MI, Pintos Morell G, Quintero Bernabeu J, et al. Síndrome de Alagille asociado a atresia intestinal. Gastroenterol Hepatol. 2016;39:667–668.
