




Wilson’s disease is a sistemic genetic disease caused by the excessive accumulation of copper. The first and main involvement is in the liver, which can range from mild and transient elevation of transaminases to the onset of an overt cirrhosis or acute liver failure. It is known that up to 20–30% of these patients may evolve to liver cirrhosis during follow-up. In clinical practice, liver fibrosis is assessed mainly by using indirect and non-invasive tools (laboratory tests, liver elastography, ultrasound), similar to other prevalent chronic liver diseases. However, despite the fact that liver elastography is a valuable tool in general hepatology, the evidence of its usefulness and accuracy in Wilson’s disease is scarce. This review summarizes the available scientific data and their limitations in Wilson’s disease.
La enfermedad de Wilson es una patología genética multiorgánica causada por la acumulación excesiva de cobre en el organismo. La afectación hepática es la inicial y principal, y puede suponer desde una hepatitis leve y transitoria, hasta el debut en forma de cirrosis o insuficiencia hepática aguda grave. En el seguimiento, hasta un 20–30% de estos pacientes evolucionan a cirrosis hepática. En la práctica clínica, la monitorización de la fibrosis hepática se realiza mayoritariamente mediante métodos indirectos y no invasivos (analíticas, elastografía hepática, ecografías) a semejanza de otras hepatopatías crónicas más prevalentes. No obstante, a pesar de que la elastografía constituye una herramienta de gran valor, la evidencia de su utilidad en Wilson es muy limitada. En esta revisión se repasa la información disponible y sus limitaciones para el seguimiento de la enfermedad de Wilson.
Wilson’s disease (WD) is a disorder of copper metabolism with an autosomal recessive inheritance pattern. It is characterised by abnormal deposition of copper in the tissues, and its signs are primarily hepatic, neuropsychiatric, corneal and haematological. The estimated prevalence of WD in the overall population is low (1/30,000); hence, it is considered a rare disease (https://www.orpha.net). However, the genetic prevalence of carriers of a pathogenic mutation in the population is approximately 1/90, suggesting that the disease could be underdiagnosed due to variable clinical penetrance.1 WD is caused by homozygous or compound heterozygous mutations in the ATP7B gene (chromosome 13), which codes for the copper-transporting protein ATP7B.2 To date, more than 1000 different pathogenic mutations have been reported (Human Gene Mutation Database [HGMD] Professional 2020.2 Release), reflecting this disease’s high level of genetic complexity.
The ATP7B protein is ubiquitous, but primary expressed in liver cells. Its role is to sequester intracellular copper to provide the substrate in the synthesis of ceruloplasmin (the main serum copper-transporting protein). It also plays a part in biliary copper excretion. Under normal conditions in a normal individual, excess copper not bound to ceruloplasmin will be excreted in faeces. Dysfunction of this ATP7B transport protein in WD results in abnormalities in the synthesis of ceruloplasmin (which is highly unstable and quickly degraded), as well as the inability to properly excrete copper in the bile, ultimately leading to accumulation of copper in toxic doses,3–5 with a great deal of potential for oxidation and cell damage in the tissues.6
The organ with the most common and earliest abnormalities in WD is the liver: WD should be considered in the differential diagnosis of any liver disease — both acute and chronic.7 Forms of hepatic presentation are highly heterogeneous and include incidental detection of hepatomegaly, fatty liver, moderate transaminase abnormalities, cirrhosis and even severe acute or fulminant hepatitis. Age of onset also varies widely. While WD has classically been thought of as a disease of young patients (<40 years of age), some patients have been diagnosed at ages that not considered characteristic of the disease (>70 years of age), suggesting the existence of clinical forms with lower penetrance.8,9 The presence of liver abnormalities associated with psychiatric and/or neurological symptoms, non-immune haemolytic anaemia or a history of the disease in first-degree relatives facilitate suspicion and diagnosis of WD. The most commonly used parameters for diagnosis are: serum ceruloplasmin levels (typically low: ≤20 mg/dl), 24-h urine copper (typically high: ≥100 mcg) and copper in dry liver tissue (typically high: ≥250 mcg/g).7,10 However, none of these parameters is powerful enough to ensure diagnosis in isolation, with variable sensitivity and specificity.11 In recent years, direct determination of exchangeable copper (CuEXC or non–ceruloplasmin-bound copper fraction) has been proposed as a highly powerful diagnostic marker, although it is not in widespread use (outside of France).12–14 In clinical practice, especially in patients with hepatic presentation and inconclusive laboratory results, a combination of different clinical and laboratory parameters compiled in the 2001 Leipzig criteria15 and endorsed by the 2012 European guidelines7 is suggested. At present, the only marker with sufficient capacity for WD diagnosis is positive genetic testing. Hepatic histology in WD is non-specific and may be indistinguishable from other types of acute or chronic liver disease.2,16 Microvesicular or macrovesicular steatosis is often seen. The only pathognomonic abnormality particular to WD is the “leaf” pattern in the mitochondrial cristae (electron microscopy), secondary to the toxic effects of copper on these organelles.16 Abnormalities in mitochondrial integrity and functioning can be detected in early stages of the disease, but this is not done in routine clinical practice.17 Histochemistry techniques for directly or indirectly visualising excess hepatic copper (rhodanine, orcein or silver staining) have low sensitivity, especially in early stages, given hepatic copper’s non-homogeneous distribution.16,18
After WD is diagnosed, early treatment is indicated, even in the absence of demonstrated tissue damage. It is focused on achieving a negative copper balance to prevent organ damage from developing or progressing. Early initiation of treatment is associated with a better prognosis for patients with WD.7 It is based on forcing urinary excretion of copper with chelating agents (d-penicillamine or trientine) or preventing intestinal absorption of copper with zinc salts.7 In cases of acute liver failure or decompensated cirrhosis that do not respond to treatment, definitive treatment with liver transplantation is indicated.2 In patients with predominantly neurological signs, treatment is based on a combination of WD treatment (prioritising low doses of chelating agents and/or zinc salts) and symptomatic treatment. Symptomatic treatment may involve oral sedatives (benzodiazepines or baclofen), intramuscular sedatives (botulinum toxin), systemic sedatives (dantrolene) or dopaminergic drugs (levodopa or dopamine agonists) as well as neuroleptics for hyperkinetic and psychiatric signs. However, neuroleptics are always preferred due to their lower risk of extrapyramidal side effects. In cases of severe generalised dystonia or dystonic storm as well as refractory tremor, deep brain stimulation of the internal globus pallidus may be considered. Neurological symptoms that do not improve after 12 months of treatment should be considered irreversible. Liver transplantation was recently proposed as an alternative treatment for WD with refractory neurological presentation.19
Follow-up of liver disease in patients with Wilson’s diseaseEven though it was first reported many years ago (1912),20 WD is still a rare disease with enormous challenges in diagnosis, follow-up and monitoring of treatment.7,21,22 Concerning hepatic manifestations, to date, there is no standardisation or consensus with regard to the medium- and long-term prospective follow-up of patients with WD. Hence, each centre acts according to its prior experience, sometimes adapting validated protocols for other more prevalent liver diseases. Generally, WD patients are followed up at hepatology clinics every 6–12 months. Published series of patients with WD have estimated that around 20%–30% of patients will eventually progress to liver cirrhosis.7,22 The factors associated with this progression are not well established, but it could be assumed that lack of adherence to medication, suboptimal treatment and the presence of comorbidities could adversely affect this unfavourable course.
Monitoring of treatment and confirmation of adherenceOne of the main difficulties in the follow-up of patients with WD involves monitoring adherence to treatment. As in other chronic diseases, adherence to long-term treatment is difficult to maintain, especially in diseases diagnosed in childhood and/or adolescence (WHO report: https://www.who.int/home). Drugs indicated for WD do not have simple dosage regimens (they require several doses per day as well as fasting to ensure absorption). Often, there often safety problems, rendering adherence a problem in many cases. Nevertheless, suitable adherence is known to be key for preventing disease progression.23,24 Lack of adherence can cause rapid worsening of the disease with serious consequences. Adherence monitoring is complex, but zinc and copper levels from 24-h urine testing can be used. In patients treated with zinc salts, urine zinc levels below 2 g or urine copper levels above 100 mcg/24 h would suggest poor adherence. In patients treated with chelating agents, urine copper levels should be kept at 200–500 mcg/24 h. Levels that are much higher or fluctuate (beyond the initial phase of treatment) are suggestive of poor adherence10 and therefore poor management of systemic copper levels.
The theoretical goal of good treatment in WD is achievement of clinical remission (reversibility of previous signs and symptoms) and biochemical response (normalisation of transaminases). However, these goals are usually not met before 6–12 months of treatment, and may be met later in patients with neurological symptoms. The clinical guidelines7,21,25 for children and adults recommend decreasing drug doses once clinical response and biochemical response have been achieved, so as not to induce excessive copper depletion with iatrogenic effects. Some patients may not show full normalisation of transaminases in follow-up, despite treatment.2 There is no clear consensus as to the timing of or algorithm for down-titration of medication in the maintenance phase of WD; however, the summaries of product characteristics for the drugs suggest not exceeding 450 mg of zinc acetate, 750 mg of d-penicillamine or 1250 mg of trientine per day (summaries of product characteristics: https://cima.aemps.es/cima/publico/home.html).
From a biochemical point of view, patient follow-up is aimed at ensuring that treatments maintain a suitable balance in copper homeostasis: namely, 24-h urine copper levels <75–100 μg (in patients treated with zinc), 200–500 μg/day (in patients on treatment with chelating agents) or non-ceruloplasmin-bound copper levels below 50−75 μg/l (in all patients). Non-ceruloplasmin-bound copper (NCC) is indirectly calculated from ceruloplasmin levels (NCC = total copper[μg/dl] — 3.15 × ceruloplasmin[mg/dl]). NCC is a biochemical marker that is difficult to calculate and highly variable, yielding results that cannot be assessed or even negative results in up to 25% of patients.10 Given its limitations, it could supplement other markers, but it could not serve as a single marker to guide clinical follow-up.
In recent years, measurement of exchangeable copper (CuEXC) has been proposed as a marker of interest.12,13 This fraction would represent free circulating copper, copper bound to other minor transport proteins (albumin or transcuprein) or copper loosely bound to ceruloplasmin (and easily interchangeable in the presence of chelating agents such as ethylenediaminetetraacetic acid [EDTA]), with the potential to cause toxicity. This experimental marker has proven useful in WD diagnosis, as well as phenotypic differentiation of symptomatic, presymptomatic and carrier patients.13 CuEXC levels are higher in patients with extrahepatic disease than in patients with isolated liver disease.14 However, to date, their usefulness in the follow-up of WD patients has not been validated.10
Thus, biochemical monitoring during follow-up is currently based on a combination of 24-h urine copper, transaminases and occasionally NCC, and it should be useful for:
- a)
Reliably determining whether a patient is adherent.
- b)
Assessing whether the drug dose is suitable, and adjusting it accordingly.
- c)
In cases of adherence and a suitable dose, considering adjusting treatment if biochemical and clinical objectives are not achieved.
In a patient with WD, one of the objectives of treatment involves normalisation of transaminases. However, this biochemical objective is not usually achieved before 6–12 months have elapsed since the start of treatment. It is important to remember that, in patients with liver impairment, chelating agents should be the first line of treatment.26 Once normalised with treatment, abnormal transaminases should prompt an investigation of whether the patient's dose and treatment adherence remain suitable, or the patient may have comorbidities or liver damage in addition to WD. The same recommendation would apply to patients in whom transaminases were not initially abnormal and to patients with changes observed in follow-up.
However, there is no significant correlation between laboratory results (transaminases) and disease severity.27,28 A typical example would be acute liver failure due to WD, which is often associated with only moderately elevated transaminase levels,2 contrasting with a marked increase in bilirubin and severe coagulopathy, which may or may not be associated with Coombs-negative haemolytic anaemia. Monitoring of hepatocellular function is essential in patients with liver cirrhosis or liver failure. The Wilson's disease prognostic index, modified by Dhawan,29 is based on markers of liver function such as albumin, aspartate aminotransferase (AST), international normalised ratio (INR), bilirubin and leukocytes; a score of more than 11 points is associated with a high probability of death on a short-term basis and is an indication for liver transplantation. In patients with no indication for liver transplantation in whom medical treatment is indicated, various measures must be implemented: treatment with copper chelating agents, dietary restrictions, avoidance of hepatotoxic agents and screening for possible complications resulting from portal hypertension (oesophageal varices and/or hepatocellular carcinoma).
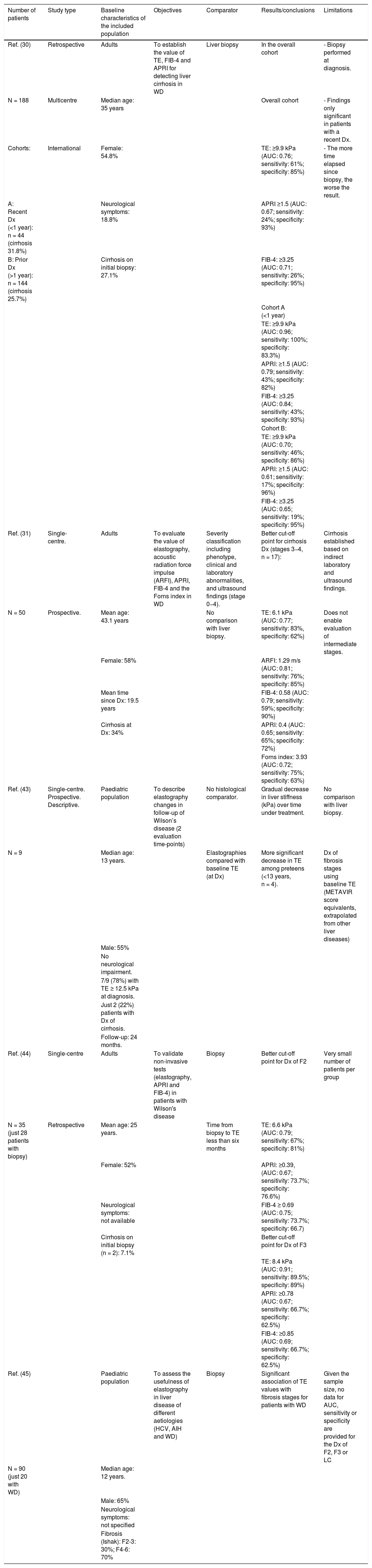
Non-invasive evaluation of liver fibrosisIn the past decade, the development of non-invasive methods and their validation in chronic liver diseases have made it possible to reduce the number of invasive procedures performed in patient follow-up. One example is transient elastography (TE), or Fibroscan, which estimates liver fibrosis by detecting wave propagation velocity.30,31 Fibroscan has been validated for monitoring fibrosis and diagnosing cirrhosis in most prevalent liver diseases: hepatitis C (12.5−14 kPa),32 hepatitis B (12.4 kPa),28,33 and non-alcoholic fatty liver disease (8.7 kPa).32 However, the use of TE in WD is more limited and there are few longitudinal studies in this regard, consistent with the low prevalence of the disease (summarised in Table 1). A recent multicentre study in central Europe34 evaluated the value of TE in 188 patients with WD and liver biopsy at diagnosis (or as an explant study). In this study, a cut-off point of 9.9 kPa was capable of accurately detecting cirrhosis (sensitivity: 100%; specificity: 83%) in recently diagnosed patients (<1-year follow-up) (n = 44; 23%). However, in the 144 patients with a longer follow-up time (1–10 years from diagnosis to Fibroscan), its accuracy was significantly lower (sensitivity: 46%; specificity: 86%). Other studies with a smaller sample size31 and heterogeneity in the gold-standard comparator have attempted to validate TE as a follow-up method in patients with WD, but have not been able to establish optimal cut-off points for monitoring fibrosis progression. While it is true that a higher Fibroscan score also appears to be correlated with a more advanced stage of liver fibrosis in these patients, the trend is towards observing values lower than those seen in other chronic liver diseases.
Review of studies with non-invasive evaluation of liver fibrosis in WD.
| Number of patients | Study type | Baseline characteristics of the included population | Objectives | Comparator | Results/conclusions | Limitations |
|---|---|---|---|---|---|---|
| Ref. (30) | Retrospective | Adults | To establish the value of TE, FIB-4 and APRI for detecting liver cirrhosis in WD | Liver biopsy | In the overall cohort | - Biopsy performed at diagnosis. |
| N = 188 | Multicentre | Median age: 35 years | Overall cohort | - Findings only significant in patients with a recent Dx. | ||
| Cohorts: | International | Female: 54.8% | TE: ≥9.9 kPa (AUC: 0.76; sensitivity: 61%; specificity: 85%) | - The more time elapsed since biopsy, the worse the result. | ||
| A: Recent Dx (<1 year): n = 44 (cirrhosis 31.8%) | Neurological symptoms: 18.8% | APRI ≥1.5 (AUC: 0.67; sensitivity: 24%; specificity: 93%) | ||||
| B: Prior Dx (>1 year): n = 144 (cirrhosis 25.7%) | Cirrhosis on initial biopsy: 27.1% | FIB-4: ≥3.25 (AUC: 0.71; sensitivity: 26%; specificity: 95%) | ||||
| Cohort A (<1 year) | ||||||
| TE: ≥9.9 kPa (AUC: 0.96; sensitivity: 100%; specificity: 83.3%) | ||||||
| APRI: ≥1.5 (AUC: 0.79; sensitivity: 43%; specificity: 82%) | ||||||
| FIB-4: ≥3.25 (AUC: 0.84; sensitivity: 43%; specificity: 93%) | ||||||
| Cohort B: | ||||||
| TE: ≥9.9 kPa (AUC: 0.70; sensitivity: 46%; specificity: 86%) | ||||||
| APRI: ≥1.5 (AUC: 0.61; sensitivity: 17%; specificity: 96%) | ||||||
| FIB-4: ≥3.25 (AUC: 0.65; sensitivity: 19%; specificity: 95%) | ||||||
| Ref. (31) | Single-centre. | Adults | To evaluate the value of elastography, acoustic radiation force impulse (ARFI), APRI, FIB-4 and the Forns index in WD | Severity classification including phenotype, clinical and laboratory abnormalities, and ultrasound findings (stage 0−4). | Better cut-off point for cirrhosis Dx (stages 3−4, n = 17): | Cirrhosis established based on indirect laboratory and ultrasound findings. |
| N = 50 | Prospective. | Mean age: 43.1 years | No comparison with liver biopsy. | TE: 6.1 kPa (AUC: 0.77; sensitivity: 83%, specificity: 62%) | Does not enable evaluation of intermediate stages. | |
| Female: 58% | ARFI: 1.29 m/s (AUC: 0.81; sensitivity: 76%; specificity: 85%) | |||||
| Mean time since Dx: 19.5 years | FIB-4: 0.58 (AUC: 0.79; sensitivity: 59%; specificity: 90%) | |||||
| Cirrhosis at Dx: 34% | APRI: 0.4 (AUC: 0.65; sensitivity: 65%; specificity: 72%) | |||||
| Forns index: 3.93 (AUC: 0.72; sensitivity: 75%; specificity: 63%) | ||||||
| Ref. (43) | Single-centre. Prospective. Descriptive. | Paediatric population | To describe elastography changes in follow-up of Wilson’s disease (2 evaluation time-points) | No histological comparator. | Gradual decrease in liver stiffness (kPa) over time under treatment. | No comparison with liver biopsy. |
| N = 9 | Median age: 13 years. | Elastographies compared with baseline TE (at Dx) | More significant decrease in TE among preteens (<13 years, n = 4). | Dx of fibrosis stages using baseline TE (METAVIR score equivalents, extrapolated from other liver diseases) | ||
| Male: 55% | ||||||
| No neurological impairment. | ||||||
| 7/9 (78%) with TE ≥ 12.5 kPa at diagnosis. | ||||||
| Just 2 (22%) patients with Dx of cirrhosis. | ||||||
| Follow-up: 24 months. | ||||||
| Ref. (44) | Single-centre | Adults | To validate non-invasive tests (elastography, APRI and FIB-4) in patients with Wilson's disease | Biopsy | Better cut-off point for Dx of F2 | Very small number of patients per group |
| N = 35 (just 28 patients with biopsy) | Retrospective | Mean age: 25 years. | Time from biopsy to TE less than six months | TE: 6.6 kPa (AUC: 0.79; sensitivity: 67%; specificity: 81%) | ||
| Female: 52% | APRI: ≥0.39, (AUC: 0.67; sensitivity: 73.7%; specificity: 76.6%) | |||||
| Neurological symptoms: not available | FIB-4 ≥ 0.69 (AUC: 0.75; sensitivity: 73.7%; specificity: 66.7) | |||||
| Cirrhosis on initial biopsy (n = 2): 7.1% | Better cut-off point for Dx of F3 | |||||
| TE: 8.4 kPa (AUC: 0.91; sensitivity: 89.5%; specificity: 89%) | ||||||
| APRI: ≥0.78 (AUC: 0.67; sensitivity: 66.7%; specificity: 62.5%) | ||||||
| FIB-4: ≥0.85 (AUC: 0.69; sensitivity: 66.7%; specificity: 62.5%) | ||||||
| Ref. (45) | Paediatric population | To assess the usefulness of elastography in liver disease of different aetiologies (HCV, AIH and WD) | Biopsy | Significant association of TE values with fibrosis stages for patients with WD | Given the sample size, no data for AUC, sensitivity or specificity are provided for the Dx of F2, F3 or LC | |
| N = 90 (just 20 with WD) | Median age: 12 years. | |||||
| Male: 65% | ||||||
| Neurological symptoms: not specified | ||||||
| Fibrosis (Ishak): F2-3: 30%; F4-6: 70% |
AIH; autoimmune hepatitis; AUC: area under the curve; Dx: diagnosis; F: fibrosis stage; HCV: hepatitis C virus; LC: liver cirrhosis; Ref.: reference; TE: transient elastography; WD: Wilson’s disease.
Other serological markers developed for purposes of non-invasive evaluation of liver fibrosis (such as AST to platelet ratio index [APRI], Fibrosis-4 [FIB-4] and the Forns index) have proven useful in other liver diseases to rule out cirrhosis, due to their high specificity, with a cut-off point of 1.5 for APRI and 3.25 for FIB-4.32 However, the usefulness of these non-invasive markers of fibrosis in patients with WD has not been reliably established to date either. A study by Paternostro et al.34 reported the association of APRI and FIB-4 with histological cirrhosis, but only in recently diagnosed patients (<1 year), in whom elastography already adequately detected cirrhosis. Other studies35 have reported an association between these markers and elastography, but with no comparative approach using liver biopsy, the value of these observations is limited.
Invasive evaluation of liver fibrosisClassically, liver fibrosis has been directly measured using liver biopsy, which constitutes the gold standard for comparison. Liver biopsy with transparietohepatic or transjugular access is an invasive procedure that is not risk-free in which a sample of liver parenchyma is obtained by puncture.36 In recent years, the development of non-invasive strategies has greatly reduced the need for liver biopsies in most patients.
Although liver biopsy is firmly accepted as part of the diagnostic algorithm for WD in both paediatric and adult patients,7,21,25,37 the usefulness of follow-up biopsies is less so. As in many other chronic liver diseases, the invasiveness inherent to biopsy has left it restricted to cases with unexplained clinical worsening or diagnostic uncertainty. However, in WD, considering the lack of specificity of the histological condition, and the limitations of the above-mentioned non-invasive techniques, follow-up biopsies in WD could have multiple objectives:
- a)
To measure copper in tissue after a variable treatment time.
- b)
To stage the disease and evaluate histological progression under treatment.
- c)
To rule out co-occurrence of additional liver diseases.
Regarding the value of measuring intrahepatic copper in patients with WD under treatment, the available evidence is limited and often old. In 1990, Gibbs et al.38 reported an association between treatment with tetrathiomolybdate and a decrease of copper in liver tissue. Other more recent studies of small series with histological follow-up over time27,39 were unable to detect an association of intrahepatic copper levels with progression or non-progression of histological disease, or with type of treatment received. Possible explanations for a lack of copper normalisation or a lack of association of intrahepatic levels with histological improvement or worsening include the great heterogeneity of copper deposition in the parenchyma, in addition to sample variability, variable monitoring time and the fact that some treatments (especially zinc salts) could lead to accumulation of non-toxic forms of copper, which would also be deposited in the liver. However, due to the small sample sizes in these studies with longitudinal evaluation of WD using biopsy, it is difficult to draw conclusions in this regard.35
Few studies have evaluated liver fibrosis using biopsy over time in patients with WD, and they have yielded mixed results and variable histopathological reports. Cope-Yokoyama et al.27 included 12 patients with WD followed up for an average of five years (range: 1–12 years), in whom they performed percutaneous follow-up biopsies (at least two: one at diagnosis and another after a mean of four years). Most cases (n = 11) were treated with the same treatment throughout the study period. Following blind examination of the biopsies by two experienced pathologists, the authors reported lesion stability or improvement in six cases (improvement of steatosis in three and improvement of fibrosis in one), but observed progression of histological lesions (inflammation and/or fibrosis) in all others (n = 6). With these data, the authors reported a rate of fibrosis of 0 fibrosis units per year in the first four years (between the first and second biopsies), which increased to 0.25 fibrosis units in three years between the second and third biopsies (performed in just four patients), but were unable to determine factors predictive of histological progression. Given the results, the authors suggested performing a liver biopsy after three years as a justifiable approach to care, even in the absence of longitudinal studies with larger sample sizes. Other studies that have evaluated histological progression in a combined total of 42 patients with WD40–45 have yielded variable results, with overall improvement over time in the parameters of fibrosis, inflammation and steatosis with biopsies 2–10 years apart. All things considered, it could be affirmed that there is not yet enough evidence to recommend follow-up liver biopsy, especially in patients with stable laboratory results, clinical findings and treatment. However, the limitations of all these non-invasive forms of follow-up are indisputable; therefore, liver biopsy should not be ruled out over time.
Finally, it must be borne in mind that patients with WD may have other liver problems in the course of their lives. It is not uncommon for these patients to have a concomitant metabolic syndrome, and alcohol use may be relevant. Liver biopsy could aid in detecting histological features of comorbidities and adjusting treatments.
Neurological follow-up of patients with Wilson’s diseaseConsidering that neurological signs in WD range from subtle, intermittent abnormalities to acute, extremely severe forms, it is advisable for all patients with WD to be assessed by neurology.
In cases with few or no symptoms, elective outpatient follow-up can be performed based on the patient’s history and clinical examination. It is advisable to perform initial brain magnetic resonance imaging, with subsequent imaging follow-up based on the patient’s clinical course and metabolic follow-up.
In symptomatic cases, apart from metabolic treatment (chelating agents and zinc salts), applicable symptomatic treatments must be used depending on neurological signs and will be closely monitored with magnetic resonance imaging, which can show reversible lesions (“face of giant panda” sign in the midbrain and signal abnormalities in the lentiform nucleus) and irreversible damage (necrotic/cystic lesions in the basal ganglia, brainstem and cerebellum). A recent study proposed plasma neurofilament levels as a biomarker of neurological impairment in WD, which could include potential usefulness in the follow-up of treatment with chelating agents.46
ConclusionsWD is a rare disease that currently faces major challenges in clinical follow-up. The scant literature, limitations of non-invasive techniques for monitoring fibrosis, difficulty of monitoring treatment and adherence due using imperfect markers, dearth of treatment options and possible co-occurrence of other liver diseases render optimal treatment of many of these patients difficult. In light of this, liver biopsy could continue to be a useful tool for both diagnosis and follow-up, especially in cases that follow an atypical or variable course.
Conflicts of interestZM is a consultant for Alexion®, Orphalan® and DeepGenomics®. YC is a consultant for Alexion®. GHQ, AD and XF declare that they have no conflicts of interest.
Please cite this article as: Herrera-Quiñones G, Dafieno AM, Compta Y, Forns X, Mariño Z. Enfermedad de Wilson: consideraciones para optimizar el seguimiento a largo plazo. Gastroenterol Hepatol. 2022;45:146–154.
