




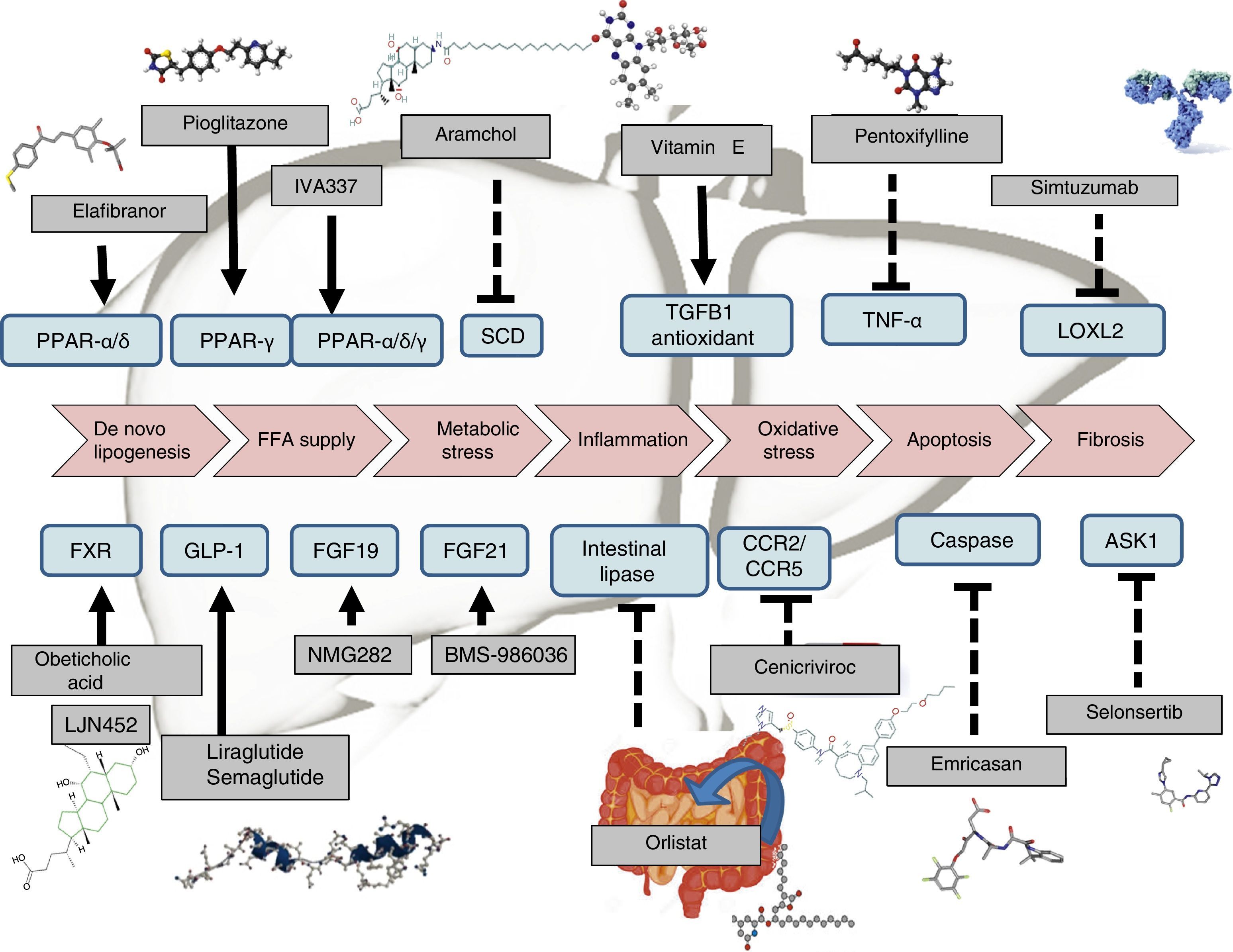
Management of non-alcoholic steatohepatitis is focused on restitution of metabolic derangement, weight loss and drugs able to improve steatosis, ballooning and fibrosis. Life-style interventions based on Mediterranean diet and increasing physical activity are the first line therapy. In patients with unsuccessful life-style intervention several drugs are under development: agonist PPAR, agonist GLP-1R and agonist FXR together with drugs focussing on inflammation, ballooning, apoptosis and fibrosis. Bariatric surgery or advanced endoscopy are reserved for morbid obese without response to life-style intervention and weighting loss drugs.
El manejo terapéutico de la esteatohepatitis no alcohólica se basa en la restitución de las alteraciones metabólicas, la pérdida de peso y los fármacos capaces de mejorar la esteatosis, la inflamación, la degeneración balonizante y la fibrosis. Las intervenciones sobre el estilo de vida basadas en dieta mediterránea e incremento de la actividad física configuran la primera línea del manejo de esta enfermedad. En pacientes en los que fracasa la intervención de estilo de vida se han de utilizar fármacos. Existen numerosos fármacos en desarrollo, entre los que destacan los agonistas FXR, los agonistas PPAR y los agonistas GLP-1R. Terapias dirigidas a las diferentes lesiones histológicas están también en desarrollo para mejorar la esteatosis, la inflamación, la degeneración balonizante, la apoptosis y la fibrosis. La cirugía bariátrica y la endoscopia terapéutica avanzada de la obesidad están reservadas a pacientes con obesidad mórbida en los que fracasan todas las opciones terapéuticas disponibles.
The importance of non-alcoholic fatty liver disease (NAFLD) has increased considerably over the last few years, to such an extent that it is now the most prevalent liver disease and one of the main causes of liver transplant in the Western world.1 It is estimated that NAFLD is detected in between 20% and 30% of the population. The prevalence of NAFLD gradually increases with age and is higher in men than in women (these differences are more pronounced in individuals under the age of 50).2 Since it is a highly prevalent disease, there are many more patients with benign disease with little impact on prognosis; however, the small percentage of patients with more advanced forms of disease account for the primary cause of liver disease in the Western world. There is an increased risk of liver disease progression in patients with non-alcoholic steatohepatitis, especially in those with fibrosis; there is also an increased risk of cardiovascular disorders and solid tumours. Therefore, treatment is indicated in those patients at risk of disease progression (steatohepatitis, fibrosis, age >60 years, male or presence of type 2 diabetes mellitus). NAFLD has 4 associated phenotypes: obesity, diabetes, metabolic syndrome and NAFLD in metabolically healthy non-obese patients. In brief, there is significant overlap between the disease pathways seen in NAFLD. NAFLD is triggered by a high influx of free fatty acids to the liver, which is especially pronounced in obesity due to insulin resistance (both in liver and in muscle and adipose tissue). NAFLD also sees an increase in de novo lipogenesis, especially due to hyperinsulinaemia, which is characteristic of metabolic syndrome and type 2 diabetes mellitus, resulting in insufficient secretion of triglycerides in the form of very-low-density lipoproteins (VLDL) and fat accumulation. Lipotoxicity is responsible for oxidative stress, with increased production of oxygen-derived free radicals, and endoplasmic reticulum stress, triggering mechanisms of apoptosis, impaired immune response and hepatic stellate cell activation which induces fibrosis progression. These pathogenic phenomena occur in the context of interactions between the patient's exposome, microbiome and genome. Gut microbiota can affect the liver through a number of mechanisms: a) intestinal barrier dysfunction, which allows bacterial translocation and the migration of bacterial products such as lipopolysaccharides; b) impaired metabolism of short-chain fatty acids and intestinal alcohol production; c) impaired metabolism of bile acids, causing gut microbiota to inhibit primary bile acid synthesis in the liver by inhibiting the farnesoid X receptor (FXR).3
Therapeutic recommendations in patients in whom lifestyle interventions have failed have been designed to tackle the patient's obesity, diabetes or metabolic disorders. Drugs are also being designed to directly treat steatosis, steatohepatitis (inflammation and ballooning degeneration) and fibrosis (Table 1). Drugs that have shown beneficial effects are pioglitazone and vitamin E, but both the FDA and the EMA do not currently recommend any drug therapy and the AASLD-EASL-EASD guidelines only recommend the use of such drugs for patients with a histological diagnosis of non-alcoholic, non-diabetic and non-cirrhotic steatohepatitis.15
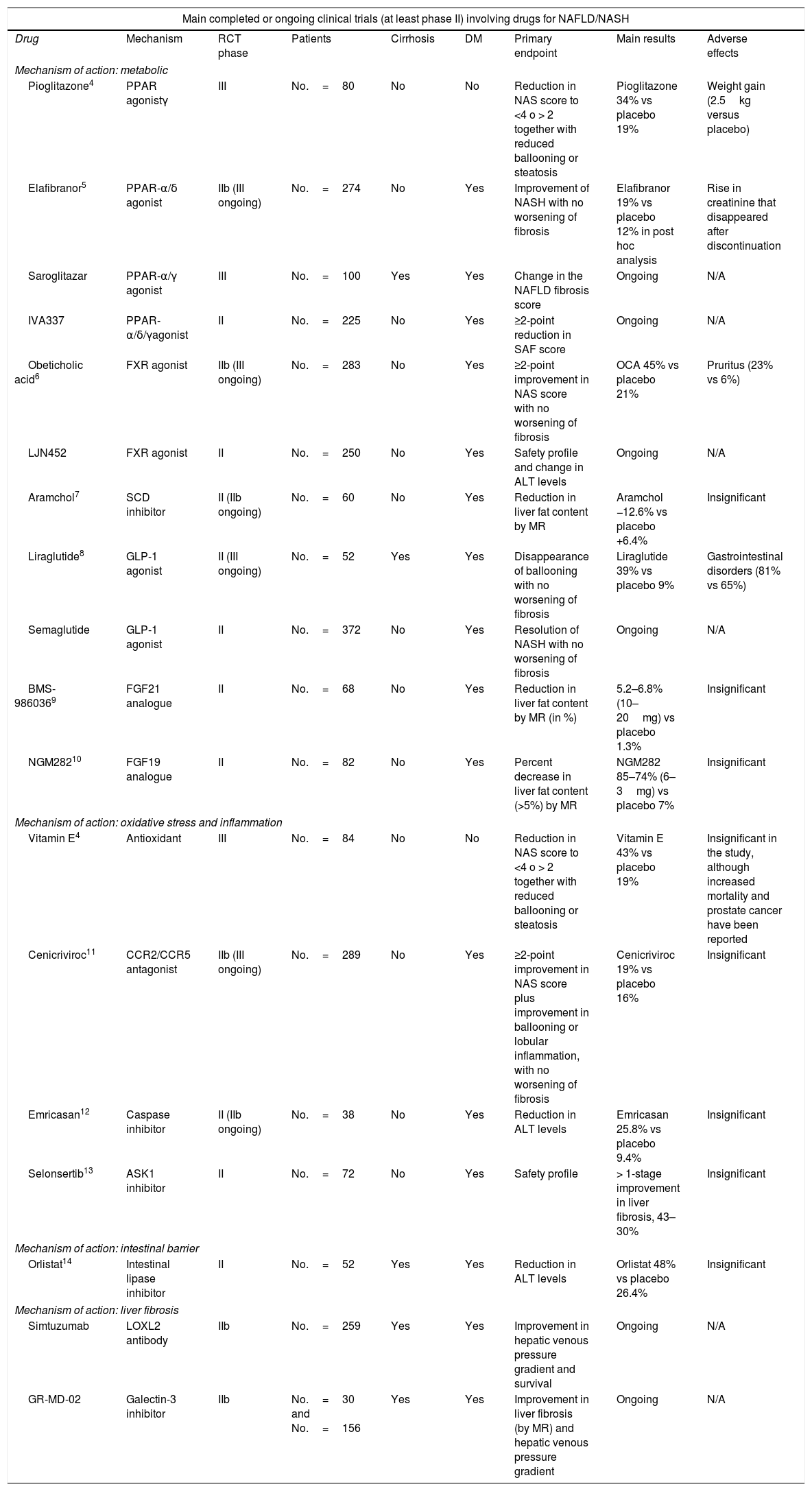
Main randomised, placebo-controlled clinical trials in patients with NAFLD/NASH.
| Main completed or ongoing clinical trials (at least phase II) involving drugs for NAFLD/NASH | ||||||||
|---|---|---|---|---|---|---|---|---|
| Drug | Mechanism | RCT phase | Patients | Cirrhosis | DM | Primary endpoint | Main results | Adverse effects |
| Mechanism of action: metabolic | ||||||||
| Pioglitazone4 | PPAR agonistγ | III | No.=80 | No | No | Reduction in NAS score to <4 o > 2 together with reduced ballooning or steatosis | Pioglitazone 34% vs placebo 19% | Weight gain (2.5kg versus placebo) |
| Elafibranor5 | PPAR-α/δ agonist | IIb (III ongoing) | No.=274 | No | Yes | Improvement of NASH with no worsening of fibrosis | Elafibranor 19% vs placebo 12% in post hoc analysis | Rise in creatinine that disappeared after discontinuation |
| Saroglitazar | PPAR-α/γ agonist | III | No.=100 | Yes | Yes | Change in the NAFLD fibrosis score | Ongoing | N/A |
| IVA337 | PPAR-α/δ/γagonist | II | No.=225 | No | Yes | ≥2-point reduction in SAF score | Ongoing | N/A |
| Obeticholic acid6 | FXR agonist | IIb (III ongoing) | No.=283 | No | Yes | ≥2-point improvement in NAS score with no worsening of fibrosis | OCA 45% vs placebo 21% | Pruritus (23% vs 6%) |
| LJN452 | FXR agonist | II | No.=250 | No | Yes | Safety profile and change in ALT levels | Ongoing | N/A |
| Aramchol7 | SCD inhibitor | II (IIb ongoing) | No.=60 | No | Yes | Reduction in liver fat content by MR | Aramchol −12.6% vs placebo +6.4% | Insignificant |
| Liraglutide8 | GLP-1 agonist | II (III ongoing) | No.=52 | Yes | Yes | Disappearance of ballooning with no worsening of fibrosis | Liraglutide 39% vs placebo 9% | Gastrointestinal disorders (81% vs 65%) |
| Semaglutide | GLP-1 agonist | II | No.=372 | No | Yes | Resolution of NASH with no worsening of fibrosis | Ongoing | N/A |
| BMS-9860369 | FGF21 analogue | II | No.=68 | No | Yes | Reduction in liver fat content by MR (in %) | 5.2–6.8% (10–20mg) vs placebo 1.3% | Insignificant |
| NGM28210 | FGF19 analogue | II | No.=82 | No | Yes | Percent decrease in liver fat content (>5%) by MR | NGM282 85–74% (6–3mg) vs placebo 7% | Insignificant |
| Mechanism of action: oxidative stress and inflammation | ||||||||
| Vitamin E4 | Antioxidant | III | No.=84 | No | No | Reduction in NAS score to <4 o > 2 together with reduced ballooning or steatosis | Vitamin E 43% vs placebo 19% | Insignificant in the study, although increased mortality and prostate cancer have been reported |
| Cenicriviroc11 | CCR2/CCR5 antagonist | IIb (III ongoing) | No.=289 | No | Yes | ≥2-point improvement in NAS score plus improvement in ballooning or lobular inflammation, with no worsening of fibrosis | Cenicriviroc 19% vs placebo 16% | Insignificant |
| Emricasan12 | Caspase inhibitor | II (IIb ongoing) | No.=38 | No | Yes | Reduction in ALT levels | Emricasan 25.8% vs placebo 9.4% | Insignificant |
| Selonsertib13 | ASK1 inhibitor | II | No.=72 | No | Yes | Safety profile | > 1-stage improvement in liver fibrosis, 43–30% | Insignificant |
| Mechanism of action: intestinal barrier | ||||||||
| Orlistat14 | Intestinal lipase inhibitor | II | No.=52 | Yes | Yes | Reduction in ALT levels | Orlistat 48% vs placebo 26.4% | Insignificant |
| Mechanism of action: liver fibrosis | ||||||||
| Simtuzumab | LOXL2 antibody | IIb | No.=259 | Yes | Yes | Improvement in hepatic venous pressure gradient and survival | Ongoing | N/A |
| GR-MD-02 | Galectin-3 inhibitor | IIb | No.=30 and No.=156 | Yes | Yes | Improvement in liver fibrosis (by MR) and hepatic venous pressure gradient | Ongoing | N/A |
Finally, there is a growing interest in this disease, as can be seen from the number of reviews published over recent months on the drug and non-drug treatment of non-alcoholic steatohepatitis.16–21
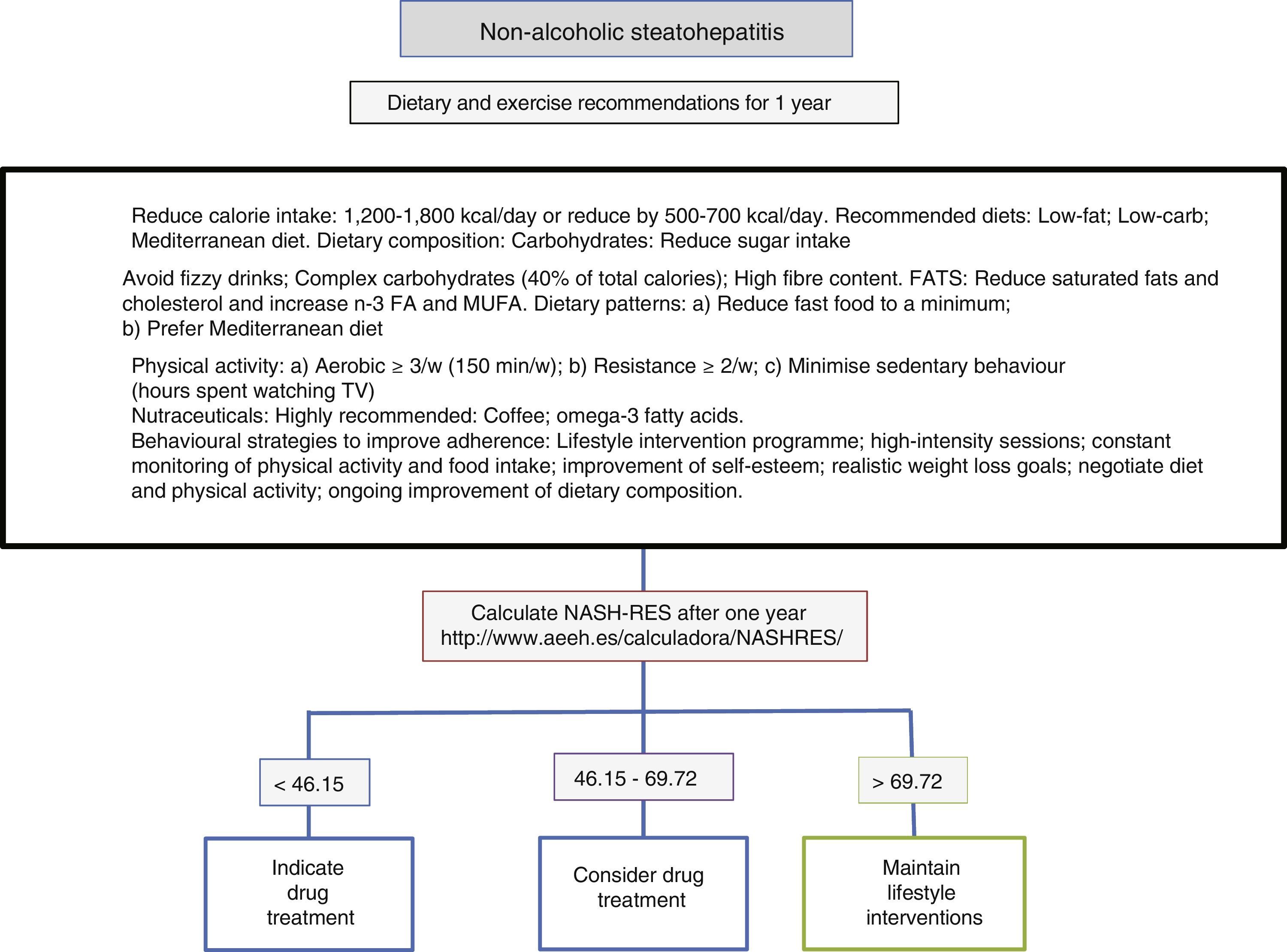
Diet, physical activity and exercise for the management of non-alcoholic steatohepatitisThe first line of treatment involves a change in lifestyle to promote a Mediterranean diet, avoid sedentary behaviour and increase physical activity and moderate aerobic exercise in order to induce weight loss, which in turn improves the patient's liver disease (Fig. 1). A low-calorie diet promotes weight loss, but no differences have been observed between a low-fat and a low-carb diet. In patients following isocaloric diets, ingestion of monounsaturated (such as olive oil) or polyunsaturated fatty acids (such as sunflower oil) promotes the elimination of fat accumulated in the liver (assessed by magnetic resonance spectroscopy). The Mediterranean diet which is characterised by a high intake of olive oil, fibre, nuts, omega-3-rich fish oil, fruits and vegetables and a low intake of refined sugars, saturated fats and processed red meat prevents the development of hepatic steatosis and steatohepatitis.22 However, a low-calorie, low-fat diet with a reduction of 500kcal/day (<30%) has been shown to induce resolution of steatohepatitis and regression of fibrosis in patients with weight loss, and, therefore, the percentage of patients showing improvement is directly proportional to the percentage of weight lost. Sedentary behaviour (assessed as the number of hours spent watching television without a break) and spontaneous physical activity (assessed as inactive, minimally active or moderately active) must be assessed properly for each patient in order to prescribe the best physical exercise (defined as planned activity in the form of aerobic, resistance, high-intensity intermittent or vigorous-intensity aerobic exercise) and the most appropriate diet. The main elements involve avoiding prolonged sedentary behaviour (a break is recommended at least every hour), avoiding physical inactivity and promoting moderate-intensity aerobic physical exercise with high-intensity bursts.23 No differences have been found between moderate aerobic exercise and resistance exercise.24 In one prospective study involving 293 patients with biopsy-proven non-alcoholic steatohepatitis, a low-fat, low-calorie diet, at least 200min of physical activity per week, a physical activity questionnaire and group behavioural sessions to promote diet adherence were prescribed. In those patients with weight loss ≥10%, 90% had resolution of non-alcoholic steatohepatitis, and 81% had fibrosis regression of at least one stage.25 A subsequent analysis in 261 patients with paired liver biopsies showed that transaminase normalisation (<19 in women and <30 in men) and the percentage of weight loss, age, baseline presence of diabetes and NAS score >5 made it possible to calculate the probability of steatohepatitis resolution (NASH-RES).26 Patients with a score of less than 46.12 should be considered for drug treatment, since the probability of disease resolution with this strategy is almost nil. However, those patients with a score of more than 69.75 should continue with the same attitude and be considered free of risks for disease progression.
Drug therapy using lifestyle interventions and drug treatment in patients with no NASH resolution options.")
Extensive research is currently being done on the development of therapies for NAFLD. Certain drugs are already fairly advanced in terms of their development, and are therefore closer to being marketed. Many other therapies, however, only have preliminary results or are currently being tested in phase II or phase III clinical trials (Table 1). Drugs can generally be classified according to their mechanism of action: a) metabolism, including peroxisome proliferator-activated receptor (PPAR),4 FXR,6 fibroblast growth factor (FGF)9,10 and glucagon-like peptide-1 (GLP-1)8 receptor agonists; b) oxidative stress and inflammation, including vitamin E,4 cenicriviroc,27 emricasan12 and selonsertib13; c) liver fibrosis, such as simtuzumab or GR-MD-02. These latter drugs will be indicated in patients with cirrhosis of the liver. In fact, they are currently being tested in clinical trials to evaluate their effect on hepatic venous pressure gradient and survival.
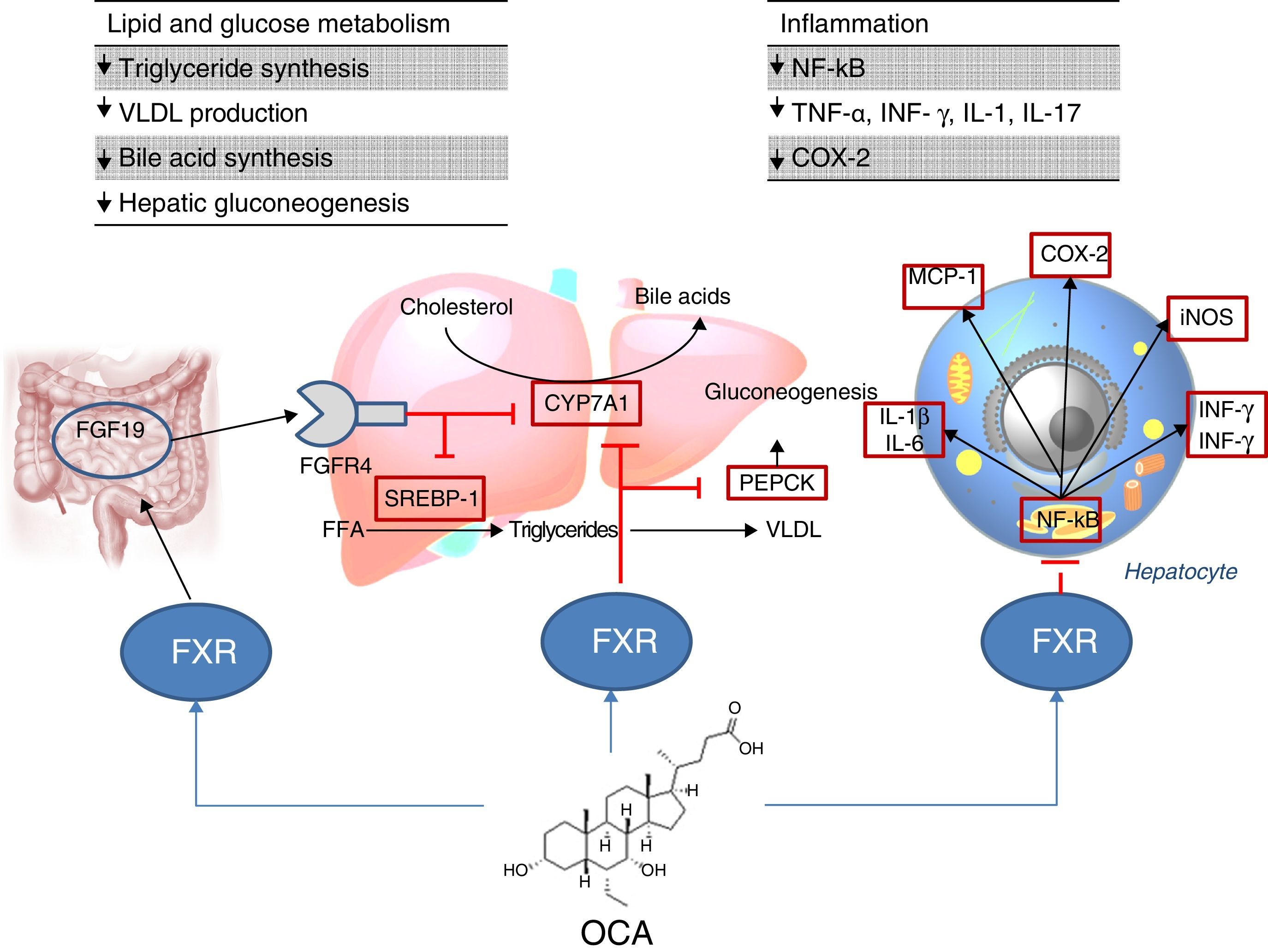
Site of action: metabolismFXR agonistsFXR activation regulates cholesterol metabolism (Fig. 2).28 Once bile acids are formed from cholesterol, these are stored in the gallbladder from where they flow into the small intestine when food is ingested; 95% of bile acids are recycled by the enterohepatic circulation. To make up for the 5% of bile acids lost from the enterohepatic circulation, the liver synthesises bile acids again, which may cause cell toxicity due to accumulation in the liver. To prevent this, bile acids bind to FXR, suppressing the expression of cholesterol 7α-hydroxylase (CYP7A1) and reducing new synthesis. However, FXR induces the expression of FGF15/19 in enterocytes, activating the JNK signalling pathway (which also decreases CYP7A1 gene expression).29 With regard to glucose and lipid metabolism, FXR has been shown to play an important role in the regulation of phosphoenolpyruvate carboxykinase (PEPCK) gene expression and other genes involved in lipoprotein metabolism.30 Various studies in FXR knockout mice (FXR –/–) have shown: a) increased levels of glucose and circulating free fatty acids (developing insulin resistance and hepatic steatosis)31; b) increased levels of HDL (reducing the expression of genes involved in reverse cholesterol transport) and non-HDL cholesterol, and apolipoprotein B-containing lipoprotein synthesis and intestinal cholesterol absorption30; c) lower serum triglyceride levels after decreasing hepatic expression of SREBP-1c.32 FXR also has a hepatic anti-inflammatory action by suppressing nuclear factor κB (NFκB).33 FXR activation also reduces expression of proinflammatory cytokines in macrophages (induced by lipopolysaccharides), which suggests it directly reduces inflammatory response in immune cells.34

Obeticholic acid (OCA) (6-ethylchenodeoxycholic acid) is a semi-synthetic derivative of chenodeoxycholic acid (CDCA) and is more potent than CDCA35; it shows FXR agonistic activity. The pharmacokinetic profile of OCA is well established. It is conjugated with glycine or taurine in the liver and is then secreted into bile. These conjugates: a) are absorbed in the small intestine where they undergo enterohepatic circulation; b) are deconjugated in the ileum and colon by gut microbiota; c) release the acid, which can be reabsorbed or excreted in faeces, the main route of elimination (<3% is excreted in urine). Both OCA and its conjugates circulate primarily (>99%) bound to plasma proteins. According to the European Medicines Agency,36 maximum plasma concentrations of the drug are reached within 2h, and are not affected by administration with food. Given that OCA is metabolised in the liver, its plasma concentrations are increased in patients with moderate and severe hepatic impairment, and a reduced dose is recommended in these cases. In individuals with renal impairment (glomerular filtration rate up to 50ml/min/1.73m2), no negative impact has been demonstrated, although its effects on the presence of advanced renal impairment are unknown.
The FLINT study analysed the role of OCA in NAFLD6; this is a randomised, multicentre, double-blind, parallel-group, placebo-controlled clinical trial in patients with non-cirrhotic, non-alcoholic steatohepatitis. The aim was to evaluate treatment with oral OCA (25mg per day) for 72 weeks. A 45% histological improvement (defined as a ≥2-point improvement in NAS score without worsening of fibrosis) was observed in patients with OCA compared to 21% in the placebo group. Improvement in hepatic steatosis, inflammation, hepatocyte ballooning and fibrosis and a decrease in transaminase levels were also observed in treated patients. However, the proportion of patients with steatohepatitis resolution was not significantly different between the two groups. Another phase II clinical trial evaluated the impact of OCA on insulin sensitivity in patients with NAFLD and type 2 diabetes mellitus, revealing a 24% improvement in treated patients versus a 5% deterioration in those patients receiving placebo.37
With regard to adverse effects, patients receiving OCA showed an increase in total and LDL cholesterol (0.38mmol/l [95% CI: 0.16–0.60], p=0.0009; 0.45mmol/l [95% CI: 0.26–0.65], p=0.0001, respectively) and a decrease in HDL (−0.06mmol/l [95% CI: −0.10 to 0.01], p=0.01). More clinical trials need to be conducted to confirm whether this atherogenic profile represents an increased cardiovascular risk. On the other hand, the drug was well tolerated, although the incidence of pruritus was higher with OCA than with placebo (23% vs 6%, p<0.0001)6 (also reported in primary biliary cholangitis),38 with this increase being dose dependent.39 After discontinuing the drug, abnormal lipid profile values returned to pre-treatment levels and pruritus disappeared. Kidney function has been evaluated in a post hoc analysis of the FLINT study,40 in both OCA and placebo patients. A decrease in glomerular filtration rate has been observed in patients treated with OCA compared with the placebo group (−1.9±7.8ml/min/year versus −0.2±7.2ml/min/year; p=0.02). It is therefore recommended that kidney function be monitored in patients with steatohepatitis treated with OCA.
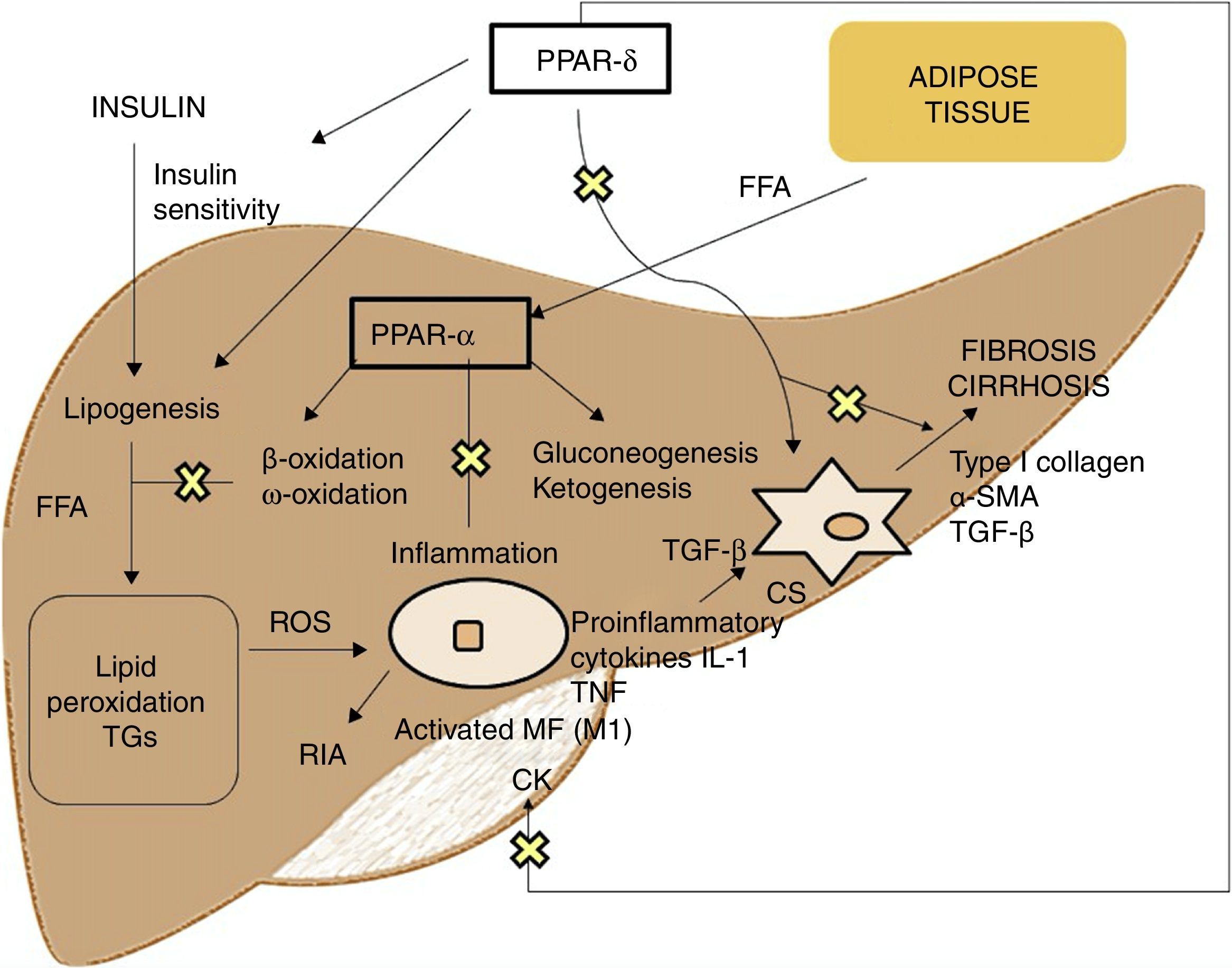
PPAR agonistsPPARs act as transcription factors and regulate energy homeostasis, inflammatory response and lipid and glucose metabolism41 (Fig. 3). Based on homology, 3 PPAR isotypes have been identified. PPAR-α, located on chromosome 22q12–13.1, is fundamentally expressed in the liver and brown adipose tissue, as well as in macrophages and parenchymal cells.42 PPAR-δ, located on chromosome 6p21.2–21.1, is expressed ubiquitously in different tissues.43 Finally, PPAR-γ, located on chromosome 3p25, is highly expressed in adipose tissue with low levels in the liver.44

PPAR-α activation regulates inflammatory response through modulation of IL-6, which results in suppression of the acute-phase response proteins stimulated by IL-6, such as serum amyloid A, α2-macroglobulin and plasminogen.45 It also has a regulatory effect on the expression of anti-inflammatory genes such as IL-1ra and IkBα, a cytoplasmic NF-kB inhibitor that plays a fundamental role in immune and inflammatory response.46 PPAR-δ activation improves insulin sensitivity, increases fatty acid transport and oxidation, decreases lipogenesis and has anti-inflammatory activity on macrophages and Kupffer cells.47 The benefit of PPAR-δ on glucose homeostasis is based on the induction of signalling molecules in adipose tissue which indirectly improves glucose removal from the muscle; such molecules include TNFα, plasminogen and leptin, which interact with insulin signalling pathways, and also free fatty acids (FFA), which determine muscle insulin sensitivity. Agonists of this receptor enhance the expression of genes involved in FFA metabolism and the uptake of these by adipocytes, decreasing their availability in muscle and improving muscle insulin sensitivity.48 Its activation in white adipose tissue also stimulates the expression of genes involved in fatty acid uptake and storage, including lipoprotein lipase, CD36/FAT, aP2 (fatty acid-binding protein) and phosphoenolpyruvate carboxykinase. Thus, the activation of PPARs causes sequestration of lipids into white adipose tissue, for storage, or into the liver for oxidation. These actions reduce the amount of lipids available to skeletal muscle, where they may otherwise interfere with insulin action.49 With regard to lipid metabolism, it promotes cholesterol efflux by inducing ABCA1 (reverse cholesterol transporter), inhibits intestinal cholesterol absorption by negatively regulating the Niemann-Pick C1-like gene, stimulates fatty acid oxidation and use by increasing the expression of target genes that control these metabolic pathways and regulates lipogenesis by inducing the expression of the insulin-induced gene (Insig-1) resulting in the suppression of key enzymes in fatty acid biosynthesis, such as SREBP-1.50 It also promotes adipocyte differentiation and redistribution of fats from the liver and muscle into the adipose tissue, preventing intracellular accumulation of fatty acids and their metabolites (fatty acyl-CoA, diacylglycerol and ceramides). In macrophages, it attenuates the production of proinflammatory cytokines (IL-1β, MIP-1α and ICAM-1) through ERK1/2 and AKT/FoxO-dependent signalling pathways51 and regulates macrophage and Kupffer cell activation. In this sense, PPAR-δ plays a key role in the balance between M1 (with proinflammatory potential) and M2 (with anti-inflammatory potential) liver macrophage phenotypes so that activation of the receptor promotes polarisation from M1 to M2, resulting in a more pronounced anti-inflammatory activity. One well-known mechanism that controls inflammatory response is negative interference with proinflammatory signalling pathways such as AP-1, NF-kB or STAT-3 in activated M1 macrophages.52 Also, PPAR-δ reduces and prevents liver fibrosis as it plays a role in the activation, phenotypic alteration and maintenance of the quiescent phase of hepatic stellate cells, also suppressing the production of type I collagen, α-SMA and TGF-β. This receptor can block TGF-β signalling pathways and Smad-dependent promoting activity, antagonising the activation and/or function of Smad3 in fibroblasts.53
Elafibranor (2-[2,6-dimethyl-4-[(1E)-3-[4-(methylthio)phenyl]-3-oxo-1-propen-1-yl]phenoxy]-2-methylpropanoic acid), with its active metabolite (GFT1007), is a dual PPAR-α and PPAR-δ agonist, with greater activity at PPAR-α (EC50: 45nmol/l for GFT505 and 15nmol/l for GFT1007) than PPAR-δ (EC50: 175 and 75nmol/l, respectively), but no activity at PPAR-γ.54 It is excreted primarily into the bile, and is largely reabsorbed into the enterohepatic circulation.55 Elafibranor has been evaluated in NAFLD in various studies since 2011. Cariou et al.5 published two studies on its use in obese patients (with dyslipidaemia or prediabetes, respectively), showing its beneficial effect for the first time. In both studies, the drug reduced plasma triglyceride levels and increased HDL and improved insulin sensitivity (decrease in HOMA-IR) as well as liver function markers. There were no major adverse events that limited the study, although a moderate increase in creatinine was observed that was reversible after discontinuation of the drug. In 2013, Cariou et al.54 conducted a clinical trial in obese, insulin-resistant patients in whom a state of hyperinsulinaemia–euglycaemia was induced by infusing insulin and non-radioactive isotopically-labelled glucose. Compared with placebo, elafibranor was shown to improve liver, muscle and adipose tissue insulin sensitivity, and also improved transaminase levels. In 2016, Ratziu et al.56 conducted a phase IIb clinical trial in which patients with non-cirrhotic steatohepatitis were randomised to receive elafibranor 80mg, 120mg or placebo for one year. A higher proportion of patients in the 120mg dose group experienced histological resolution of steatohepatitis without worsening of fibrosis and an improvement in NAS score than the placebo group. Improvements in histological score were accompanied by a change in liver function markers and non-invasive methods for fibrosis. Once again the drug was well tolerated, although a slight increase in creatinine was observed (reversible after discontinuation of the drug).
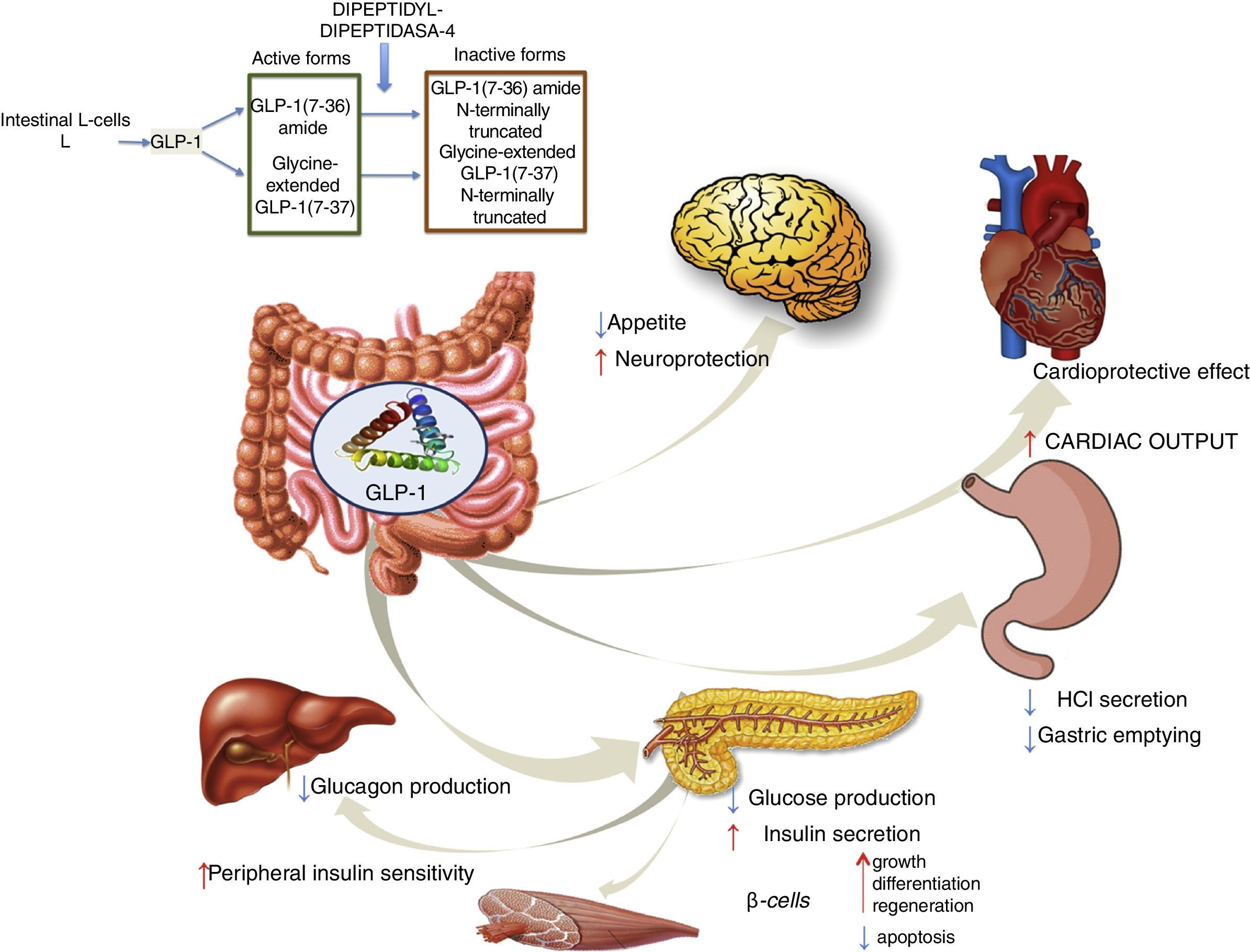
GLP-1 receptor agonistsGLP-1 is an intestinal incretin hormone that is secreted in both healthy individuals and in patients with type 2 diabetes after ingesting high-sugar and high-fat food.57 GLP-1 triggers a pancreatic response, increasing insulin synthesis and inhibiting glucagon synthesis. GLP-1 has demonstrated effects on different targets: a) on pancreatic β cells, promoting their growth, differentiation and regeneration while inhibiting apoptosis; b) in the stomach, reducing gastric acid secretion and slowing gastric emptying58; c) in the brain, reducing appetite and increasing neuroprotection; d) it reduces hepatic glucose production; e) it improves peripheral insulin sensitivity; f) and it shows cardioprotective effects by increasing cardiac output (Fig. 4). GLP-1 analogues have been shown to reduce liver enzyme and oxidative stress levels, improving histological behaviour. A direct action on liver cells has also been shown, reducing lipogenesis and increasing fatty acid oxidation.59

Liraglutide is a long-acting GLP-1 analogue that was approved for glycaemic control in overweight patients with type 2 diabetes mellitus. Endogenous GLP-1 peptides are degraded in just a few minutes by the dipeptidyl peptidase-4 (DPP-4) enzyme, while liraglutide is metabolised much more slowly. The LEAN (Liraglutide Efficacy and Action in NASH) study8 is a multicentre, double-blind, randomised trial that evaluated the effects of liraglutide versus placebo after 48 weeks in patients with non-alcoholic steatohepatitis, defined as macrovesicular steatosis (>5%) and hepatocyte ballooning, presence of Mallory's hyaline identified by immunohistochemistry and lobular inflammation, plus a BMI>25kg/m2. Patients with type 2 diabetes were selected if they had stable glycaemic control (glycated haemoglobin<9%) and were managed by either diet or stable dose of metformin or sulfonylurea. The primary endpoint of the study was histological improvement, defined as disappearance of steatohepatitis with no worsening of fibrosis. A total of 52 patients were recruited who were randomised to two groups, which received liraglutide and placebo, respectively. Finally, 23 patients from the study drug group and 22 from the placebo group had biopsies at baseline and 48 weeks after starting treatment. Nine (39%) of the 23 patients in the liraglutide group versus 2 (9%) of the 22 patients in the placebo group had complete resolution of steatohepatitis with no worsening of fibrosis. Also, 3 (38%) of 8 patients with type 2 diabetes and 6 (40%) of 15 patients without type 2 diabetes achieved the primary endpoint with liraglutide. Neither of the 2 patients in the placebo group who obtained histological improvement had type 2 diabetes. Data on improvements in weight and glycaemic levels with liraglutide were obtained as secondary endpoints as these could have a favourable effect on the risk of developing cardiovascular disease and premature death. However, more studies with long-term outcomes are required to confirm this hypothesis. With regard to adverse effects, liraglutide was safe and well tolerated, regardless of the severity of the underlying disease.
DPP-4 inhibitorsInhibitors of the DPP-4 enzyme (responsible for degradation of endogenous GLP-1), such as sitagliptin and vildagliptin, extend the half-life and improve the activity of this peptide. Nevertheless, two randomised studies comparing the safety and efficacy of sitagliptin and placebo showed no differences in efficacy (improvement of steatosis, NAS score or fibrosis) versus placebo. One of the studies included 12 patients and the other included 50 prediabetic or diabetic patients with NAFLD.60,61 As a result, the use of DPP-4 inhibitors is not recommended for the treatment of non-alcoholic steatohepatitis.
AramcholAramchol is a synthetic mixed acid (fatty acid-bile acid) that is obtained by conjugating cholic acid (bile acid) and arachidic acid (saturated fatty acid). Its function is to inhibit activity of stearoyl-coenzyme A desaturase 1 (SCD1), the enzyme responsible for catalysing the synthesis of monounsaturated fats from fatty acids in the liver and other tissues.21 This produces a reduction in the triglyceride and fatty acid ester store in the liver, which has been seen to produce a reduction in steatosis and an improvement in insulin resistance in animal models.62 Greater SCD1 activity has been detected in obese subjects with steatohepatitis compared with obese subjects with a healthy liver. Aramchol has been evaluated in a double-blind,7 placebo-controlled clinical trial involving 60 patients with biopsy-proven NAFLD (although only 6 had steatohepatitis) who were randomised to groups of 20 patients who received 100mg or 300mg of aramchol or placebo. Aramchol at a dose of 300mg was safe and effective in reducing liver fat content as measured by magnetic resonance spectroscopy after 12 weeks of treatment. Treatment with aramchol was also associated with an increase in adiponectin levels. Phase III studies showing the effects of treatment on biopsy-proven liver injury will define the role of aramchol in therapy in the near future.
Site of action: oxidative stress and inflammationCenicriviroc is a dual CCR2 and CCR5 antagonist. These receptors are involved in inflammation and fibrosis mechanisms in steatohepatitis. CCR2/CCR5 are involved in the migration and infiltration of proinflammatory monocytes into the liver, promoting liver damage via Kupffer cell activation.63 Animal studies have demonstrated the efficacy of the drug. Cenicriviroc has been evaluated in the CENTAUR clinical trial (drug vs placebo for one year), which involved 289 patients. Its aim was to show the effect of the drug by way of a >2-point improvement in NAS score with at least a one-point improvement in ballooning or lobular inflammation with no worsening of fibrosis. The preliminary results of the CENTAUR trial showed no differences in the primary endpoint (19% vs 16%), although there was a greater improvement in liver fibrosis with cenicriviroc (20% vs 10%), especially among those patients whose NAS score was >5 (24% vs 10%). Cenicriviroc was also able to reduce levels of inflammation markers, such as IL-1β, IL-6 and CRP.11
Caspases are enzymes that play a central role in apoptosis and inflammation, playing an essential role in NAFLD pathogenesis. Emricasan is a potent, irreversible pan-caspase inhibitor. A phase I clinical trial showed that emricasan was able to improve inflammatory biomarkers in patients with liver disease.64 In a phase II clinical trial, ALT levels were significantly lower in treated patients than in patients receiving placebo.12 A late-phase trial is currently in progress to evaluate improvement in liver histology.
Apoptosis signal-regulating kinase 1 (ASK1) is a protein that plays a role in apoptosis under conditions of oxidative stress.65 Selonsertib is an ASK1 inhibitor that has been developed for the treatment of steatohepatitis. In a phase II clinical trial, 43% of the patients treated with selonsertib had a one or more stage reduction in hepatic fibrosis versus 30% of patients in the placebo group.13 An ongoing clinical trial has been designed to evaluate the efficacy of selonsertib 6mg vs 18mg, with or without simtuzumab.
Site of action: intestinal barrierOrlistat is a gastrointestinal lipase inhibitor that enhances weight loss, decreases the flow of free fatty acids to the liver and improves insulin sensitivity without producing hepatotoxic adverse effects. In a randomised, double-blind, placebo-controlled clinical trial evaluating the efficacy of orlistat in NAFLD, a total of 52 patients were randomised to receive orlistat (120mg 3 times a day) or placebo for 6 months. Patients receiving orlistat were observed to have a higher reduction in ALT and insulin levels and a more significant reversal of hepatic steatosis detected by ultrasound, an effect that is beyond that expected from weight reduction alone.66 In a series of 10 cases, Harrison et al.14 demonstrated an improvement in liver histology in patients with weight loss; steatosis improved in 6/10 patients and fibrosis improved in 3/10 patients after 6 months of treatment. Treatment with orlistat, together with dietary advice, therefore induces weight loss and improves hepatic and metabolic parameters. Administration of this drug has been associated with mainly gastrointestinal adverse effects, such as oily stools, urgent need to defaecate, diarrhoea, flatulence, abdominal pain or anal fissures; cases of cholelithiasis, acute pancreatitis and, in very few cases, acute cholestatic hepatitis and subacute liver failure have been reported. Comorbidities, such as hypertension, diabetic ketoacidosis and nervous system and renal impairment, have also been reported. Due to the above issues (safety profile and no added value beyond weight loss), its use has been limited in the context of NAFLD.67
Site of action: liver fibrosisLiver fibrosis is one of the main targets of NAFLD treatment, both to prevent its onset and to be able to reverse it if it does arise. However, very few drugs are currently being studied. The most noteworthy drug is simtuzumab, which is a monoclonal antibody against lysyl oxidase-like 2 (LOXL2), a pro-fibrotic enzyme (activates TGF-β) that is abundant in hepatocytes. In vitro studies have shown the efficacy of simtuzumab in the reduction and regression of liver fibrosis.68 This drug has already shown interesting results in primary sclerosing cholangitis.69 Clinical trials are currently being conducted to evaluate the safety and efficacy of simtuzumab in patients with and without cirrhosis with NAFLD.
Other mechanisms of actionOmega-3 fatty acidsNon-alcoholic steatohepatitis is associated with a deficiency of polyunsaturated fatty acids,70,71 which are derived from essential fatty acids and consist of molecules with high biological activity. The administration of omega-3 fatty acids produces a clear improvement in blood triglyceride levels and optimises peripheral insulin sensitivity, while reducing proinflammatory effects through different mechanisms of action.72–74 A randomised, double-blind, placebo-controlled clinical trial was conducted to analyse the role of ethyl-eicosapentaenoic acid in the treatment of non-alcoholic steatohepatitis.75 The trial included 243 patients who were randomised to 3 groups, one receiving placebo and the other 2 receiving different doses of the drug (1800 and 2700mg/dl, respectively); the results of 171 patients were finally analysed. No significant differences were found between the 3 groups with regard to the primary study endpoint (decrease in NAS score with no worsening of fibrosis) or in the subgroup analysis based on the presence or absence of diabetes. Likewise, no differences in secondary study endpoints (improvement of steatosis, lobular inflammation, hepatocyte ballooning and fibrosis) were observed. A meta-analysis of randomised, placebo-controlled trials evaluating the use of omega-3 polyunsaturated fatty acids in NAFLD has recently been published; its endpoint was a change in ALT, AST, GGT, cholesterol and triglyceride levels. The authors concluded that such treatment is effective in these patients as it decreases ALT, cholesterol and particularly triglyceride levels, and increases HDL cholesterol. However, no improvement in liver fibrosis was observed (evaluated using serum levels of type IV collagen).76 Therefore, more studies are required to assess the efficacy of this therapy in slowing progression of NAFLD.
OthersNew therapeutic targets, with alternative mechanisms of action, are being investigated. These include amylin analogues (pramlintide, davalintide), leptin analogues (metreleptin), GLP-1 agonists (exenatide, TTP054), MC4R agonists (RM-493), oxyntomodulin analogues, neuropeptide Y antagonists (velneperit), cannabinoid type 1 receptor blockers (AM6545), MetAP2 inhibitors (beloranib), lipase inhibitors (cetilistat) and vaccines to prevent obesity (ghrelin, somatostatin and Ad36), which will allow for more personalised treatment of obesity77 (Fig. 5).
Surgical and endoscopic therapy
Endoscopic therapy for non-alcoholic steatohepatitis is reserved for those patients who do not manage to lose weight by changing their lifestyle or using drugs that enhance weight loss, such as liraglutide. There is a high prevalence of advanced liver fibrosis (15%) and steatohepatitis (25%) in morbidly obese patients.78 Bariatric surgery has been shown to be beneficial in non-alcoholic steatohepatitis and fibrosis and also in other risk factors, such as diabetes mellitus,79 insulin resistance80 and hypertension.81 It can even increase GLP-1 secretion82 (associated with insulin sensitivity) and, therefore, its beneficial effects may go beyond weight loss or improved metabolic syndrome. Both steatosis and non-alcoholic steatohepatitis are reversed after bariatric surgery,83 making this a cost-effective option.84 Paired biopsy studies have shown an improvement in liver histology, including ballooning, lobular inflammation and fibrosis, one year after surgery. More specifically, steatosis and lobular inflammation can be resolved in up to 75%, portal inflammation in up to 50% and steatohepatitis in up to 90% of cases within 3 years of surgery. Patients with grade 2 (59%) and grade 3 (29%) fibrosis can also improve significantly.85 Bower et al. conducted a systematic review of 29 studies involving obese patients who had undergone bariatric surgery. The authors showed how the presence of steatosis, inflammation, ballooning and fibrosis decreased after surgery, and also revealed a significant improvement in ALT, AST and GGT values.86 Nevertheless, although the published studies indicate that bariatric surgery is associated with a significant improvement in patients with steatohepatitis, more clinical trials need to be conducted with an appropriate sample size due to the small size of the studies conducted to date and the heterogeneity of the study population. It would be interesting to determine whether the beneficial effects of bariatric surgery depend on the type of surgery performed. Most of the studies published to date are based on Roux-en-Y gastric bypass surgery. In the most important comparative study conducted to date (including paired biopsies), Mathurin et al. included 381 patients who underwent restrictive (gastric banding) and malabsorptive (Roux-en-Y gastric bypass, bilio-intestinal bypass) surgery with repeat biopsies 1 and 5 years after the procedure. The endpoint was a similar improvement in histological parameters to previous studies (steatohepatitis from 27% to 14%), observing that this improvement occurred especially during the first year. On comparing the different techniques, no differences were observed between the different procedures.87 This lack of differences has been observed in other studies.88,89 Therefore, we do not currently have any robust data to support the recommendation of any specific type of bariatric surgery in patients with NAFLD; the technique should therefore be selected based on its complexity, risk-benefit ratio and size of the excluded gastrointestinal component.
Endoscopic therapy for obesity has not yet been evaluated in detail in patients with NAFLD. One of the few studies published to date was conducted by Lee et al. to evaluate the role of an intragastric balloon (plus diet and exercise) compared with diet and exercise alone in 18 obese patients for 6 months. Those patients with intragastric balloon placement experienced greater weight loss and greater reduction in NAS score.90 Despite these promising data, it is necessary to evaluate this type of surgery in larger cohorts with longer follow-up times.
ConclusionsTreatment of non-alcoholic steatohepatitis includes a broad spectrum of therapeutic options ranging from measures aimed at modifying lifestyle, physical activity, diet and sport to the design of new molecules that are able to alter the pathogenic mechanisms of the disease. Future treatments for non-alcoholic steatohepatitis are based on PPAR, FXR and GLP-1R agonists. Molecules aimed at treating apoptosis, antioxidants and molecules aimed at treating immune response and fibrosis will fill a gap in the therapeutic algorithm for this disease over the coming years. Finally, these investigational drugs have different targets and some of them are complementary, which means that, in the future, combined treatments may improve results reported to date.
Conflict of interestNone.
Please cite this article as: Ampuero J, Sánchez-Torrijos Y, Aguilera V, Bellido F, Romero-Gómez M. Nuevas perspectivas terapéuticas en la esteatohepatitis no alcohólica. Gastroenterol Hepatol. 2018;41:128–142.
