





Fundamento: El retinoblastoma, cáncer intraocular infantil más frecuente, se presenta como esporádico (unilateral o bilateral) o afectando a varios miembros de una familia. En los casos hereditarios aparece por una mutación germinal transmitida o surgida de novo, mientras que en los no hereditarios se debe a dos mutaciones somáticas en una célula de la retina. El presente trabajo se planteó con objeto de analizar desde el punto de vista genético el gran número de familias con algún miembro afectado de retinoblastoma, recopiladas en los últimos años, para ahondar en los mecanismos moleculares que inciden en este proceso patológico y ofrecerles consejo genético.
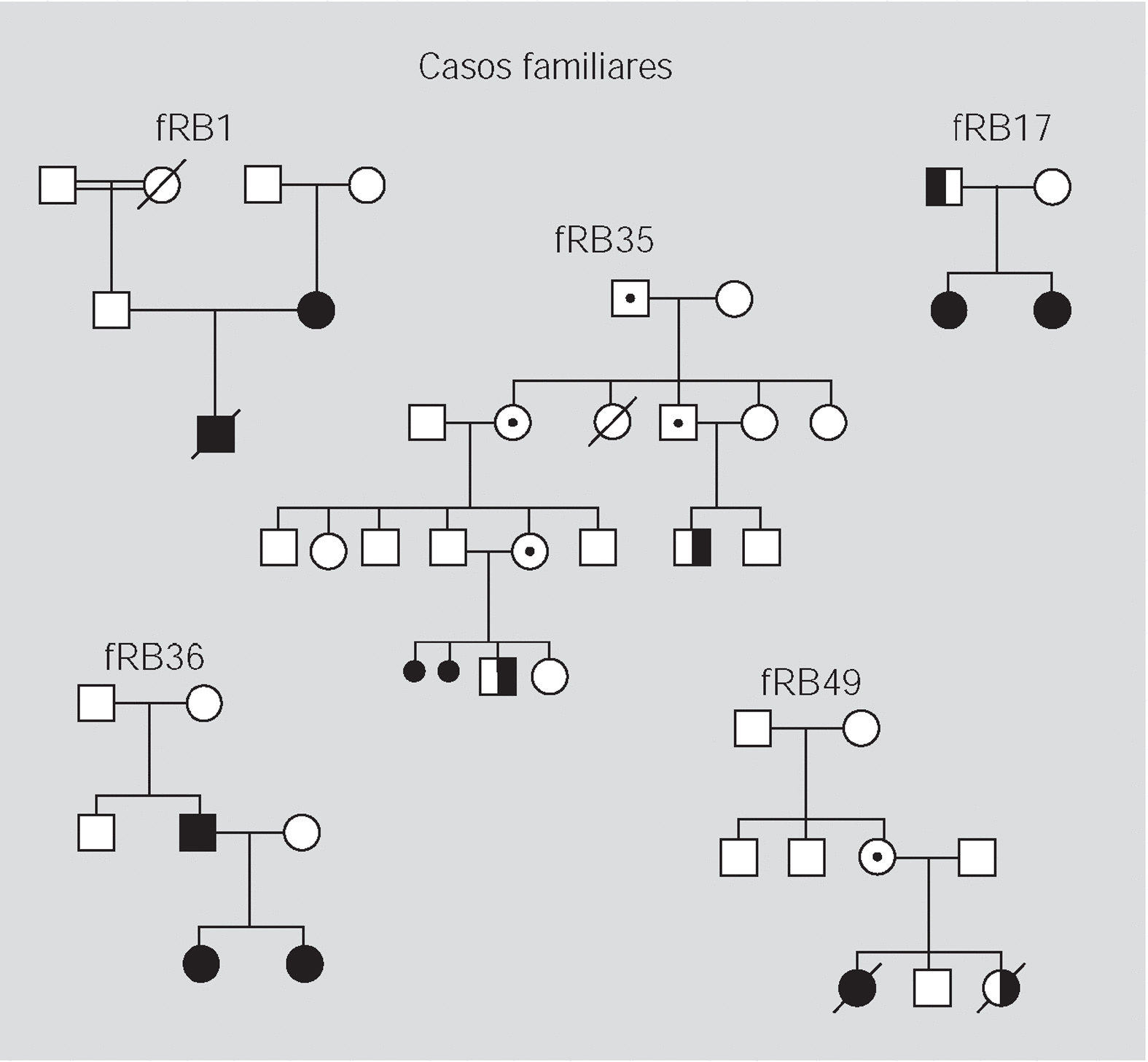
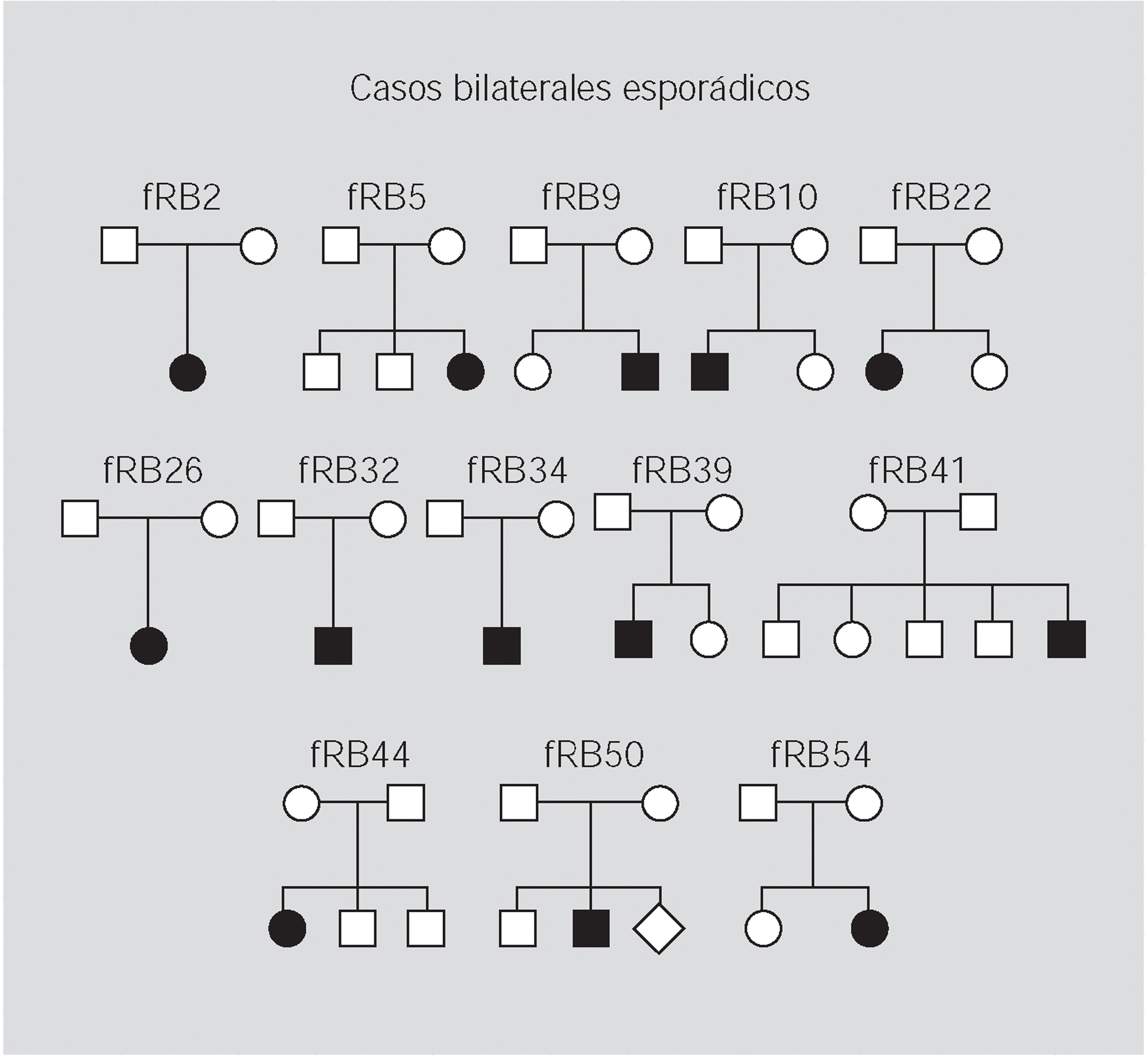
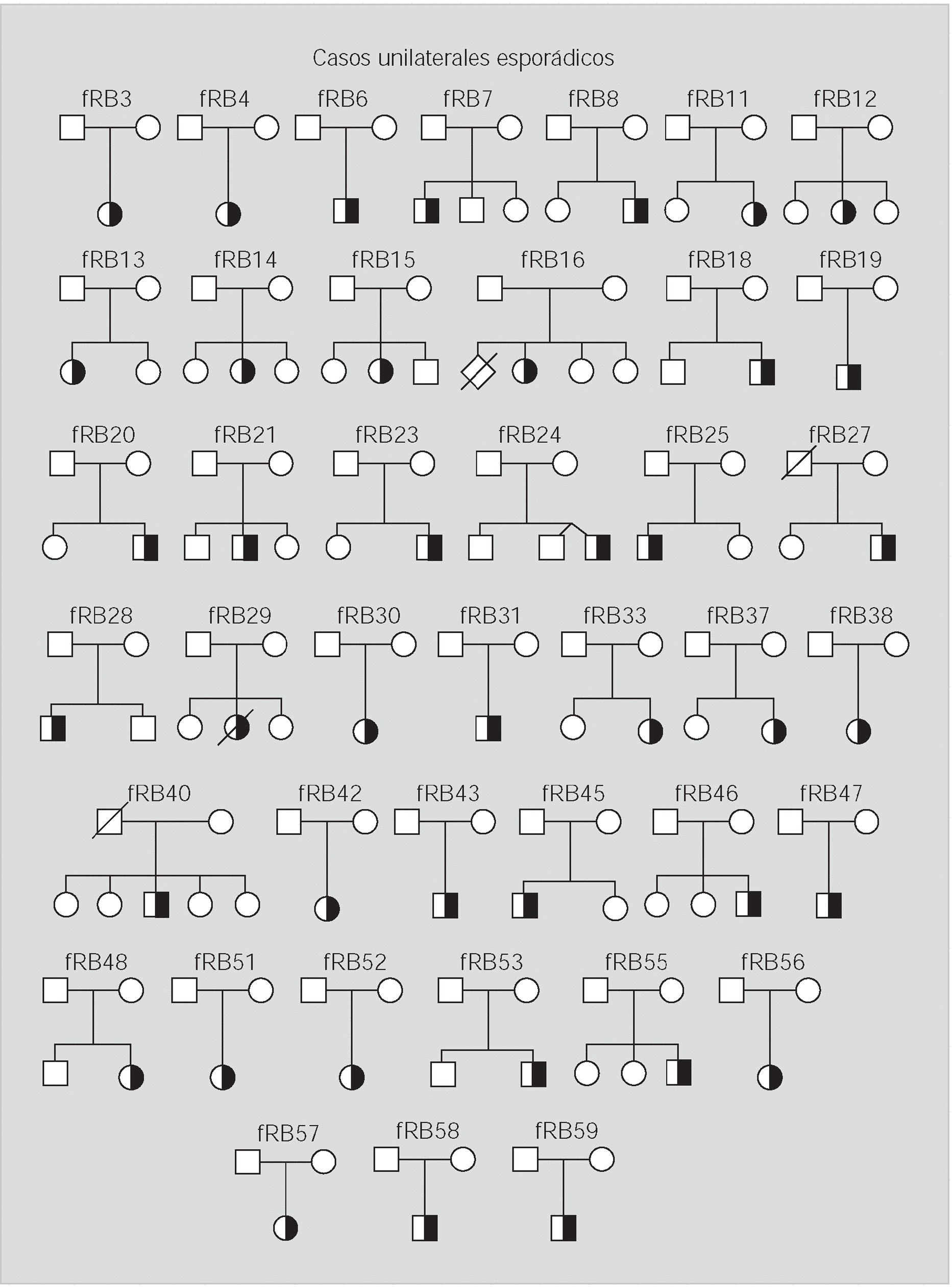
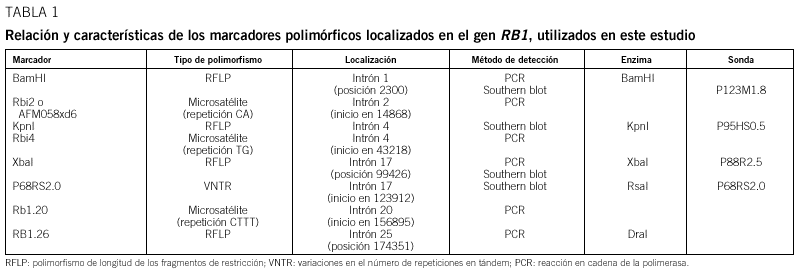
Pacientes y método: Se han analizado 59 familias con algún miembro afectado de retinoblastoma y se ha realizado un estudio citogenético, con marcadores polimórficos y de búsqueda de mutaciones en el ADN de leucocitos y de los tumores disponibles.
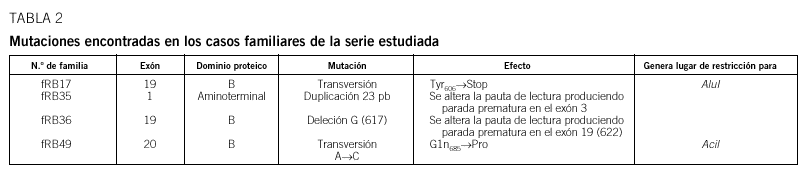
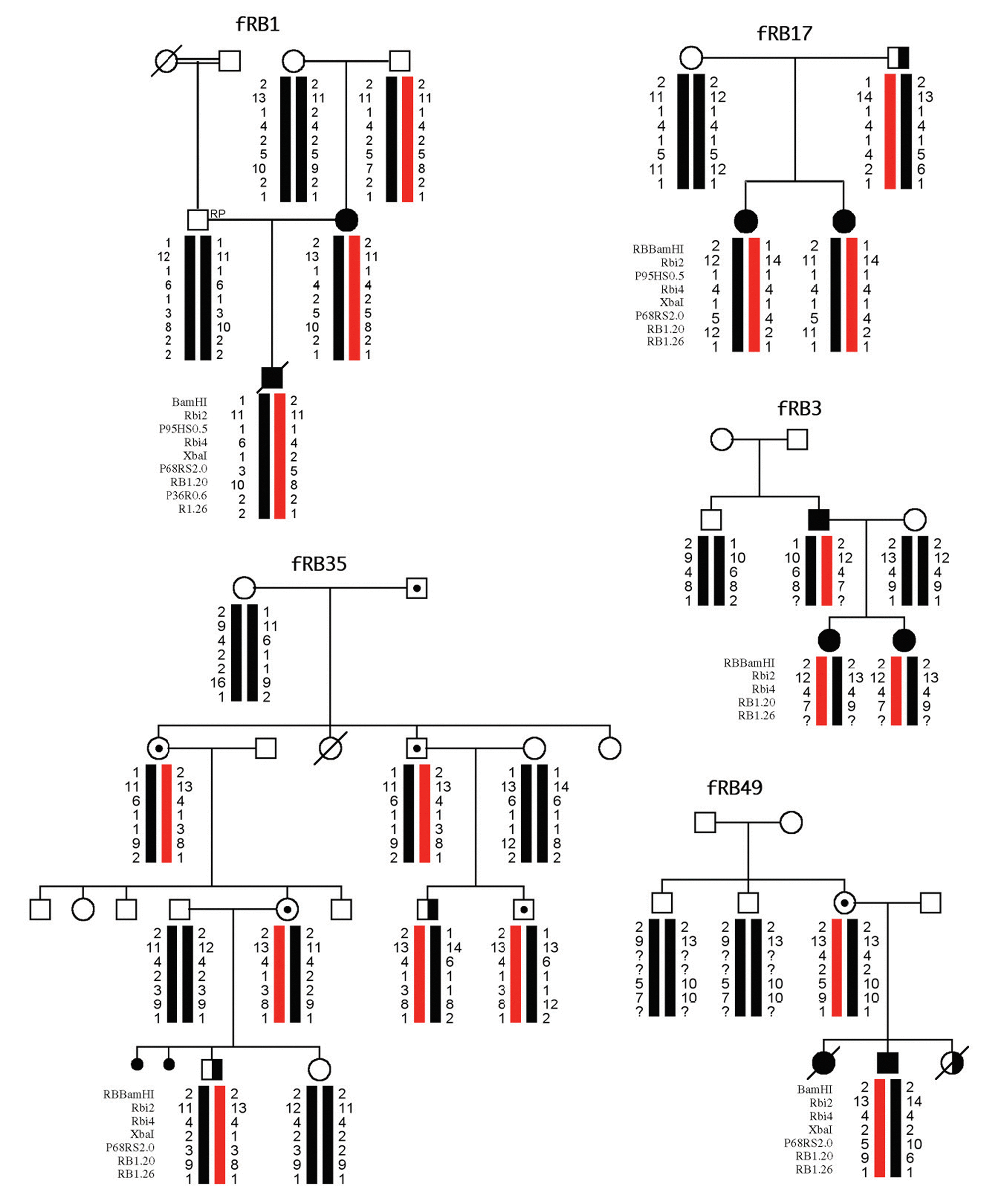
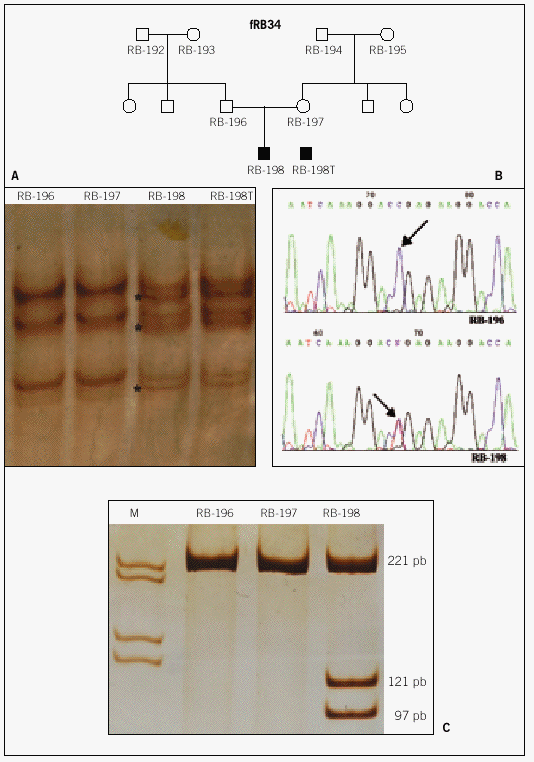
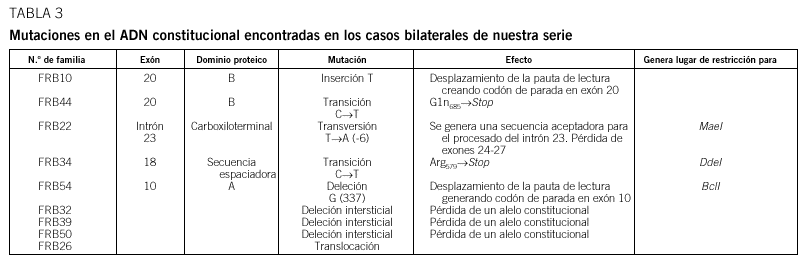
Resultados: En 4 de los 5 casos familiares, se ha establecido la mutación asociada a la enfermedad, al igual que en 9 de los 13 casos bilaterales esporádicos. En un 7% de los casos unilaterales esporádicos la mutación se encontraba en el ADN leucocitario. La pérdida de heterocigosidad como segundo episodio mutacional se debe principalmente a la recombinación mitótica.
Conclusiones: Entre las mutaciones contabilizadas en nuestra serie, se observa mayor frecuencia de mutaciones puntuales de carácter constitucional, que dan origen al primer acontecimiento mutacional, mientras que la pérdida de heterocigosidad es el episodio mutacional detectado mayoritariamente en tumores.
Background: Retinoblastoma, the intraocular malignancy most common in children, occurs in both familial and sporadic (bilateral or unilateral). Hereditary predisposition is caused by a germ-line mutation while non-hereditary is due to two somatic mutations in a retinal cell. This work was carried out in order to analyse genetically, the high number of families with some affected member and to go deep into the molecular mechanisms responsible of this pathology.
Patients and method: 59 families with one or more affected members were analysed. Cytogenetics and with polymorphic markers studies were carried out and a search for mutations was performed in DNA from white cells and from available tumoral tissue.
Results: In four of the 5 familial cases, the responsible mutation was established, the same as in 9 of the 13 bilateral sporadic. In the 7% of the unilateral sporadic cases, mutation was found in leucocytary DNA. Lost of heterozygosity as a second mutational event was mainly due to mitotic recombination.
Conclusions: Among the mutations of our series, a higher frequency of punctual mutations, responsible of the first mutational event, was observed at constitutional level. Lost of heterozygosity was the mechanism observed in the majority of the tumours.
