




En atención primaria la orientación diagnóstica es clave en la consulta diaria. El manejo correcto de los diagnósticos diferenciales es pieza fundamental en nuestra práctica. En este artículo exponemos un caso clínico que engloba el manejo de 2 bloques fundamentales, el primero de la ictericia y el segundo de la anemia hemolítica.
Aprendiendo sobre el manejo, tratamiento y atención a la familia.
In Primary Care the initial diagnostic approach is an essential factor. The correct handling of the differential diagnosis is fundamental in our practice. We present a clinical case which involves the management of two fundamental parts; the first is the jaundice and the second is haemolytic anemia.
Learning about, the management, treatment and family care.
Con frecuencia en la consulta de atención primaria se nos plantea el diagnóstico diferencial de la ictericia, y del manejo del mismo se derivan decisiones que encaminan el correcto diagnóstico de nuestros pacientes.
En el caso clínico que vamos a exponer la paciente presentó de forma brusca un cuadro de ictericia marcada.
En el diagnóstico diferencial de los cuadros de ictericia1 la clave que nos ha de orientar en primer término son siempre los hallazgos analíticos. Si solo se observa hiperbilirrubinemia, debemos pensar en trastornos hemolíticos o hiperbilirrubinemia hereditaria. Si predomina la citólisis con pruebas de imagen normales: sospechar lesión hepatocitaria de origen vírico, tóxica, autoinmune o isquémica. Si predomina la colestasis es prioritario conocer el estado (dilatación o no) de la vía biliar intra y extrahepática.
Dentro de los trastornos hemolíticos que producen hiperbilirrubinemia el déficit de la enzima glucosa-6-fosfato deshidrogenasa (G6PD) da lugar al favismo. Este se caracteriza por la aparición de forma brusca de episodios de hemólisis provocados por medicamentos, infecciones o alimentos (estrés oxidativo)2.
Las manifestaciones clínicas más habituales en adultos son las ocasionadas por la anemia hemolítica.
Caso clínicoSe presenta el caso de una mujer de 25 años que, sin antecedentes personales ni familiares de interés, acude a consulta de atención primaria porque desde el día anterior se encuentra nauseosa, cansada y al levantarse esa mañana se ve «amarilla». En la primera valoración llama la atención la marcada ictericia de la paciente.
En la anamnesis la paciente refiere no tomar anticonceptivos orales, no haber realizado viajes recientes, no tener prácticas de riesgo, estar bien vacunada y no ser consumidora habitual de alcohol. No ingesta de fármacos los días previos.
La exploración física es normal (sin dolor abdominal, ni megalias, ni adenopatías) salvo la reseñada ictericia.
Se solicita analítica urgente en la que destaca: serie roja hematíes 3,91 10E6/μl, hemoglobina (Hb) 11,2g/dl, hematocrito 36,1%, VCM 92,0 fl. Estudio de coagulación normal, en las determinaciones bioquímicas en suero/plasma: creatinina 0,70mg/dl, proteínas totales 8,8g/dl, bilirrubina total 8,4mg/dl, bilirrubina conjugada 0,0mg/dl (0,1-0,5), LDH 581 U/l (120,0-246,0), GPT (ALT) 12 U/L (0,0-41,0),GOT (AST) 60 U/l (0,0-31,0), alfa-amilasa 78 U/l (0,0-100,0).
Determinaciones cualitativas en orina mediante tira reactiva sin alteraciones.
A la vista de los resultados analíticos, planteamos el diagnóstico diferencial de la hiperbilirrubinemia1 que es el dato que en ese momento parece más llamativo. Se piensa que puede tratarse de una hepatitis vírica de inicio o un proceso de colestasis. Se realiza ecografía abdominal que es normal. Decidimos observación domiciliaria con analítica al día siguiente.
Al acudir de nuevo a consulta la paciente cuenta astenia y fiebre el día anterior termometrada (38°C), la ictericia es más llamativa y refiere coluria.
Se realiza de forma urgente una nueva analítica. El nuevo hemograma objetiva descenso de 2 puntos en la hemoglobina, las transaminasas siguen levemente aumentadas, pero no en ascenso y persiste el aumento de LDH.
A la vista de estos resultados se descarta causa hepática de la ictericia. Debido el aumento de LDH y descenso de hemoglobina, y con la sospecha de anemia hemolítica1 se decide el ingreso de la paciente en el hospital de referencia.
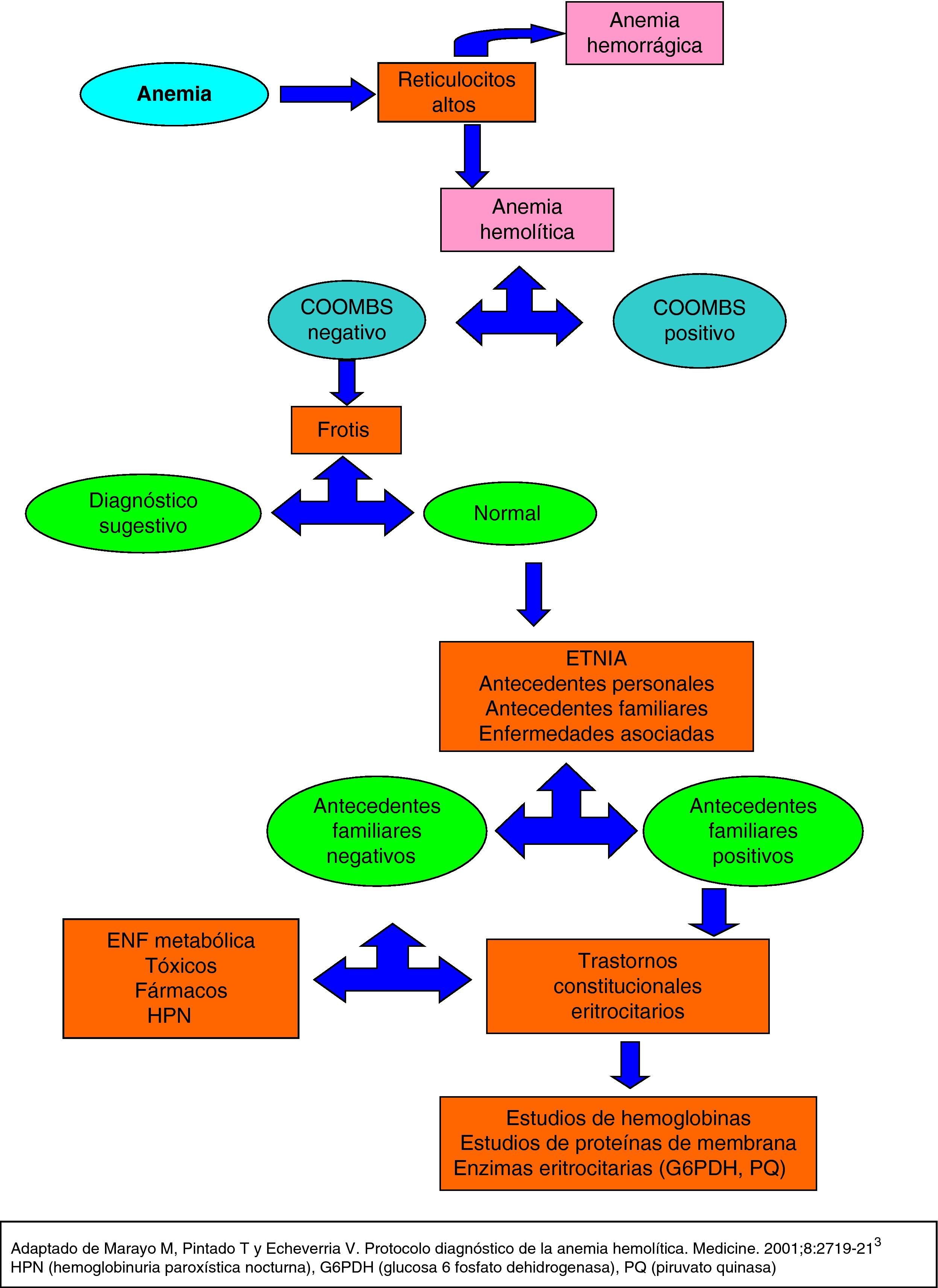
Ante la sospecha de una anemia hemolítica, primero hay que constatar la existencia de hemólisis y después esclarecer la naturaleza de la misma3.
En la práctica clínica nos guiamos por signos indirectos para deducir que la anemia es de patogenia hemolítica (fig. 1). La prueba más sencilla y útil es el recuento reticulocitario, que está habitualmente elevado evidenciando el esfuerzo compensador de la médula ósea3. La distinción entre pérdidas hemorrágicas y hemólisis no siempre es fácil; el incremento de reticulocitos puede acompañar ambas. En la anemia hemolítica hay un conjunto de parámetros bioquímicos que son muy indicativos de hemólisis: elevación de la LDH, haptoglobina descendida y aumento de la bilirrubina a expensas de la bilirrubina indirecta3.

En el ingreso la Hb desciende hasta 8,2, reticulocitos 113 (24-84) % 3,6 (0,1-2,4). LDH 844 (120-246), GOT 46, haptoglobina 9,2 (12-15).
Una vez establecida la presencia de un proceso hemolítico, las 2 pruebas de mayor utilidad son la prueba de Coombs y el estudio morfológico del frotis de sangre3.
Se realiza test de Coombs directo que al ser negativo descarta posible causa autoinmune de la hemólisis3. En la extensión de sangre periférica: se objetiva de forma aislada algún esquistocito, policromasia. No dianocitos, lleptocitos, ni dacriocitos. Serie blanca sin alteraciones. Plaquetas sin alteraciones.
En una gran parte de pacientes con anemia hemolítica no inmunológica, como es este caso, las alteraciones morfológicas eritrocitarias orientan de forma fundamental el diagnóstico.
A la vista de los resultados se decide revisar el caso, prestando especial atención a la anamnesis y la exploración.
Reinterrogada la paciente comenta que 2 días anteriores al inicio de la ictericia se llevó al trabajo para comer habas frescas, como único plato.
Según lo recogido en la anamnesis, el desencadenante de este proceso de anemia agudo podría ser la ingesta de habas orientando hacia un proceso constitucional subyacente: déficit de G6PD.
Se solicitó analítica específica en la que se detecta una cifra baja de G6PD.
En el ingreso la paciente evolucionó favorablemente con suplementos de ácido fólico y ferroterapia oral, no precisando ser transfundida.
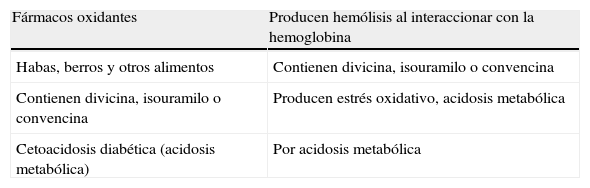
Con el diagnostico de anemia hemolítica aguda Coombs negativa debida al déficit de G6PD, se informa al alta de los fármacos y alimentos que pueden producir crisis de hemólisis (tabla 1) y se aconseja estudiar a su familia.
Factores desencadenantes
| Fármacos oxidantes | Producen hemólisis al interaccionar con la hemoglobina |
| Habas, berros y otros alimentos | Contienen divicina, isouramilo o convencina |
| Contienen divicina, isouramilo o convencina | Producen estrés oxidativo, acidosis metabólica |
| Cetoacidosis diabética (acidosis metabólica) | Por acidosis metabólica |
Fuente: Iborra J et al.1.
Las anemias hemolíticas congénitas se deben a la alteración hereditaria de la síntesis de las proteínas que constituyen la membrana del hematíe (membranopatías), de las enzimas que intervienen en su metabolismo energético (enzimopatías) o de la Hb4.
En las enzimopatías se origina perdida de la función reductora del hematíe, de forma que no puede defenderse de la acción oxidativa de determinados agentes. Se produce así la desnaturalización de la Hb, con la formación de cuerpos de Heinz y otras proteínas eritrocitarias que conducen a la disminución de la deformabilidad del hematíe y a su hemólisis inmediata4.
La enzima G6PD cataliza la primera reacción de la vía de las pentosas-fosfato, de forma que se obtiene NADPH que protege al eritrocito del estrés oxidativo, manteniendo al glutatión en estado reducido2,5.
El déficit de G6PD constituye la enzimopatía congénita más frecuente, afectando a más de 400 millones de personas en el mundo2,6, hasta un tercio de los niños que nacen con ictericia tendrán una deficiencia de G6PD6.
Su distribución geográfica es similar a la de la malaria, debido a la protección que ofrece frente a la infección por Plasmodium falciparum5,6.
Se transmite ligada al cromosoma X, teniendo las portadoras heterocigotas menor expresión clínica que los varones homocigotos. La prevalencia en España es del 1%4.
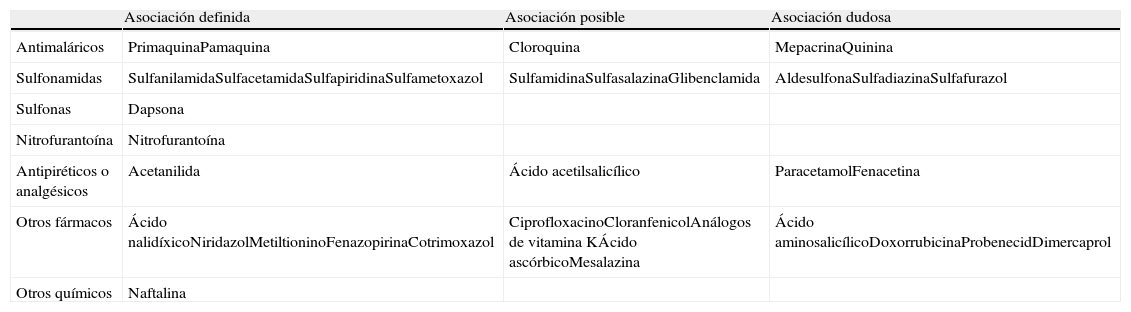
Existen más de 400 variables moleculares y 140 mutaciones descritas que dan lugar a una enzima con distinto grado de actividad y gran variabilidad clínica5. Generalmente, estos pacientes permanecen asintomáticos durante años, hasta la exposición a algún factor desencadenante (tabla 1), fundamentalmente fármacos oxidantes (tabla 2) e infecciones, con aparición de hemólisis aguda intravascular con hemoglobinuria5.
Fármacos con efecto oxidante
| Asociación definida | Asociación posible | Asociación dudosa | |
| Antimaláricos | PrimaquinaPamaquina | Cloroquina | MepacrinaQuinina |
| Sulfonamidas | SulfanilamidaSulfacetamidaSulfapiridinaSulfametoxazol | SulfamidinaSulfasalazinaGlibenclamida | AldesulfonaSulfadiazinaSulfafurazol |
| Sulfonas | Dapsona | ||
| Nitrofurantoína | Nitrofurantoína | ||
| Antipiréticos o analgésicos | Acetanilida | Ácido acetilsalicílico | ParacetamolFenacetina |
| Otros fármacos | Ácido nalidíxicoNiridazolMetiltioninoFenazopirinaCotrimoxazol | CiprofloxacinoCloranfenicolAnálogos de vitamina KÁcido ascórbicoMesalazina | Ácido aminosalicílicoDoxorrubicinaProbenecidDimercaprol |
| Otros químicos | Naftalina |
Fuente: Iborra J et al.1.
La hemólisis inducida por habas constituye un síndrome especial, conocido como favismo5, que fue descrito por Pitágoras en el siglo v7. También se relaciona con la ingesta de otros alimentos que contengan divicina, isouramilo o convecina, como los berros2.
La Organización Mundial de la salud (OMS)4,8 describe 3 variantes de la deficiencia de G6PD:
- •
Tipo 1: síndrome hemolítico crónico de intensidad variable con 0% de actividad enzimática.
- •
Tipo 2: asintomático hasta exposición a agente oxidante, con una actividad enzimática del 5-15%.
- •
Tipo 3: asintomático con actividad enzimática normal.
El diagnóstico se basa en la observación clínica y en la demostración de exposición a un factor desencadenante. Se observarán datos clínicos y analíticos de hemólisis y los característicos cuerpos de Heinz junto con anisopoiquilocitosis en el frotis de sangre periférica2.
El grado de actividad enzimática puede determinarse, de forma indirecta, mediante la medición por espectofotometría de la síntesis de NADPH o de la cantidad de glutatión reducido.
El diagnóstico definitivo requiere el estudio mutacional con análisis molecular.
La medida terapéutica más importante es evitar la exposición a los agentes desencadenantes.
La mayor parte de los pacientes permanecen asintomáticos, únicamente la exposición a uno de los agentes desencadenantes podría producir hemólisis, el grado de esta dependerá del déficit de la enzima y del grado de exposición, porque en la historia natural de la enfermedad en numerosas ocasiones hay referencia a contacto con el agente sin producir crisis graves. En caso de crisis hemolítica es necesario instaurar soporte transfusional, cobertura antibiótica, hidratación y suplementos con ácido fólico2,5.
El estudio de los familiares se hace con control analítico del nivel de enzima, se hace a los ascendientes y descendientes, comenzando por los hermanos de la persona afectada.
