






La hipercolesterolemia familiar (HF) es un trastorno genético frecuente que se manifiesta desde el nacimiento y que causa un aumento en los niveles plasmáticos de colesterol-LDL (cLDL), xantomas y enfermedad coronaria prematura. Su detección y tratamiento precoz reduce la morbimortalidad coronaria. A pesar de la disponibilidad de un tratamiento eficaz, la HF está poco diagnosticada y tratada. La identificación de los casos índices y la posterior detección en cascada familiar utilizando los niveles de cLDL y la detección genética es la estrategia más coste-efectiva para la detección de nuevos casos. El tratamiento crónico con estatinas ha disminuido el riesgo cardiovascular a los niveles de la población general. Los objetivos en cLDL son <130mg/dl en los niños y adultos jóvenes, <100mg/dl en los adultos y <70mg/dl en los adultos con enfermedad coronaria conocida o diabetes. En la mayoría de los pacientes es difícil conseguir estos objetivos, por lo que puede ser necesario el tratamiento combinado con ezetimiba u otros fármacos. Cuando no se alcanzan los objetivos con el máximo tratamiento farmacológico tolerado, una reducción de cLDL≥50% puede ser aceptable. La LDL-aféresis es útil en los pacientes homocigotos y en los heterocigotos graves resistentes al tratamiento. Este documento proporciona recomendaciones para el diagnóstico, cribado y tratamiento de la HF en niños y adultos, así como consejos específicos para los especialistas clínicos y médicos de atención primaria con el objetivo de mejorar el cuidado de los pacientes y reducir su carga de enfermedad cardiovascular.
Familial hypercholesterolemia (FH) is a common genetic disorder, clinically manifested since birth, and associated with very high levels of plasma LDL-cholesterol (LDL-c), xanthomas, and premature coronary heart disease. Its early detection and treatment reduces coronary morbidity and mortality. Despite effective treatment being available, FH is under-diagnosed and under-treated. Identification of index cases and cascade screening using LDL-c levels and genetic testing are the most cost-effective strategies for detecting new cases and starting early treatment. Long-term treatment with statins has decreased the vascular risk to the levels of the general population. LDL-c targets are <130mg/dL for children and young adults, <100mg/dL for adults, and <70mg/dL for adults with known coronary heart disease or diabetes. Most patients do not to reach these goals, and combined treatments with ezetimibe or other drugs may be necessary. When the goals are not achieved with the maximum tolerated drug treatment, a reduction ≥50% in LDL-c levels can be acceptable. Lipoprotein apheresis can be useful in homozygous, and in treatment-resistant severe heterozygous, cases. This Consensus Paper gives recommendations on the diagnosis, screening, and treatment of FH in children and adults, and specific advice to specialists and general practitioners with the objective of improving the clinical management of these patients, in order to reduce the high burden of coronary heart disease.
La hipercolesterolemia familiar (HF) es el trastorno genético más frecuente asociado con enfermedad coronaria prematura (ECP), debido a elevadas concentraciones de colesterol LDL (cLDL) desde el nacimiento1,2. Su mecanismo de transmisión es autosómico dominante y aproximadamente la mitad de la descendencia de una persona afectada presentará el trastorno. Se produce principalmente por mutaciones en el gen del receptor LDL (RLDL), y menos frecuentemente por mutaciones del gen de la apolipoproteínaB (APOB) y del gen Proprotein Convertase Subtilisin/kexin type 9 (PCSK9)1. Su prevalencia es de aproximadamente una de cada 300-500 personas en la población general, estimándose en 100.000 los españoles que padecen este trastorno1,3. El diagnóstico clínico se basa en concentraciones elevadas de cLDL, historia familiar de hipercolesterolemia, antecedentes de ECP y la presencia de xantomas y/o arco corneal1. La HF acelera la enfermedad ateroesclerótica coronaria de una a 4décadas4. En España, el 55% de los varones y el 24% de las mujeres en la década de los 50años han presentado manifestaciones de enfermedad coronaria5. La prevalencia y el elevado riesgo de desarrollar ECP hacen de la HF un problema de salud pública. A pesar del elevado riesgo cardiovascular (RCV), la mayoría de los pacientes están sin diagnosticar ni tratar. Su diagnóstico precoz permite utilizar medidas preventivas. Entre ellas, el tratamiento crónico con estatinas ha demostrado en los pacientes con HF sin enfermedad coronaria previa una marcada reducción del RCV, similar al de la población general6.
Necesidad de un documento de consenso para la detección y tratamiento de la hipercolesterolemia familiarLos pacientes con HF suelen acudir al primer nivel asistencial; sin embargo, la mayoría están sin diagnosticar y por tanto sin tratamiento, o bien con tratamiento insuficiente7,8.
En España, algunas comunidades autónomas han implementado diferentes estrategias para el diagnóstico de la HF mediante criterios clínicos y confirmación genética. Un estudio observacional español, con participación de médicos de atención primaria (AP) y especializada, ha demostrado que menos del 5% de los casos con diagnóstico genético de HF consiguen el objetivo en cLDL <100mg/dl y menos del 15% de estos están recibiendo el máximo tratamiento combinado8.
Para evitar este vacío en la prevención de la enfermedad coronaria en esta población de alto riesgo se ha elaborado este documento de consenso cuyo objetivo es revisar la información actualmente disponible acerca del diagnóstico y el tratamiento de la HF, y consensuar con un grupo de expertos recomendaciones que ayuden a los especialistas clínicos y médicos de AP a realizar un adecuado diagnóstico, tratamiento y cribado familiar con el fin de prevenir el desarrollo de la ECP. Además, este documento puede ayudar en la elaboración de políticas de prevención y promoción de la salud.
MetodologíaPara la elaboración de este documento de consenso se han seguido las recomendaciones del protocolo Appraisal of Guidelines REsearch & Evaluation (AGREE)9. La Fundación Hipercolesterolemia Familiar promovió la creación de un panel de expertos formado por 6 internistas de clínicas de lípidos, 3 cardiólogos, 2 pediatras, un médico de AP, un endocrinólogo, una investigadora en ateroesclerosis, un genetista clínico, un responsable en salud y 2 personas con HF. Se realizó una búsqueda en las bases Medline, PubMed y Cochrane de todos los temas relacionados con la HF.
Se realizaron 2 reuniones de trabajo presenciales y 2 videoconferencias en el primer semestre de 2013. Después de la revisión sistematizada de la evidencia, se discutieron y consensuaron las recomendaciones basadas en la mejor evidencia disponible, en la opinión de expertos y en la buena práctica clínica. Las recomendaciones finales se han clasificado de acuerdo a los criterios modificados del National Health and Medical Research Council10 en: a)es de confianza para guiar la práctica clínica; b)puede ser de confianza para guiar la práctica clínica en la mayoría de las situaciones; c)puede ser utilizada para guiar la práctica clínica, pero se debe tener precaución en su aplicación.
Diagnóstico clínico en adultosEl diagnóstico de la HF se basa en niveles elevados de cLDL (generalmente >220mg/dl), historia familiar de hipercolesterolemia, presencia de ECP y depósitos de colesterol en forma de xantomas y/o arco corneal (tabla 1 y fig. 1). La obtención del árbol familiar es esencial para evaluar la probabilidad de HF y para posteriormente realizar la detección familiar. Existen 3 herramientas diferentes para establecer el diagnóstico clínico de la HF en el caso índice (CI): el programa MedPed11, el Simon Broome británico12 y los criterios de la red de clínicas de lípidos holandesa (RCLH)13, siendo estos últimos los que se utilizan en España, ya que han sido validados con el diagnóstico genético que es el gold standard14,15 (tabla 2). El diagnóstico clínico es de certeza cuando la puntuación es ≥8 y de probabilidad cuando la puntuación es ≥613.
Criterios de sospecha clínica de hipercolesterolemia familiar
| 1. Individuo con cLDL >220mg/dl y al menos uno de los siguientes criterios: |
| a) Familiar < 18 años con cLDL > 150mg/dl |
| b) Familiar > 18 años con cLDL > 190mg/dl |
| c) Presencia de enfermedad coronaria prematura en el caso índice y/o en familiar de primer grado |
| d) Presencia de xantomas en el caso índice y/o en familiar de primer grado |
| 2. Si no se dispone de datos familiares se debe sospechar una hipercolesterolemia familiar en personas con cLDL > 300mg/dl |
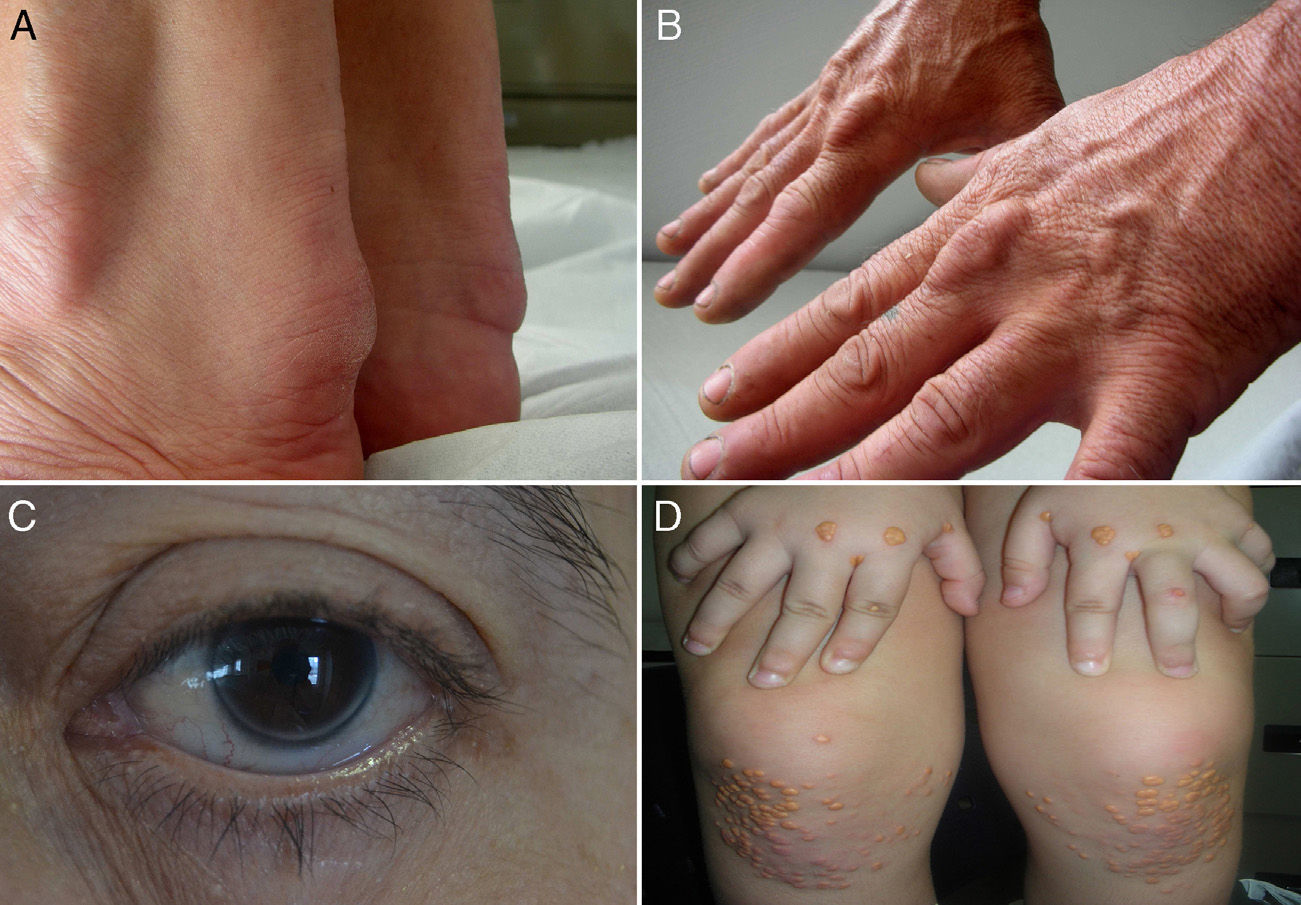
Xantomas en el tendón de Aquiles y en los extensores de la mano. C)Arco corneal completo en un varón <45años. D)Xantomas eruptivos y planos en manos y rodillas de un niño de 5años con HFHo. (Fotos cortesía de la Fundación Hipercolesterolemia Familiar.) Reproducido de Expert Review of Cardiovascular Therapy, March 2013, Vol. 11, No. 3, Pages 327-342, con permiso de Expert Reviews Ltd.")
Signos de hipercolesterolemia familiar. A, B)Xantomas en el tendón de Aquiles y en los extensores de la mano. C)Arco corneal completo en un varón <45años. D)Xantomas eruptivos y planos en manos y rodillas de un niño de 5años con HFHo. (Fotos cortesía de la Fundación Hipercolesterolemia Familiar.) Reproducido de Expert Review of Cardiovascular Therapy, March 2013, Vol. 11, No. 3, Pages 327-342, con permiso de Expert Reviews Ltd.
Criterios de la red de clínicas de lípidos holandesas para el diagnóstico de hipercolesterolemia familiar
| Historia familiar | |
| Familiar de primer grado con enfermedad coronaria prematura (hombres <55 años y mujeres < 60 años) y/o | 1 |
| Familiar de primer grado con niveles de cLDL > 210 mg/dl | |
| Familiar de primer grado con xantomas tendinosos y/o arco corneal < 45 años y/o | 2 |
| Familiar < 18 años con cLDL ≥ 150 mg/dl | |
| Antecedentes personales | |
| Paciente con enfermedad coronaria prematura (hombres < 55 años y mujeres < 60 años) | 2 |
| Paciente con enfermedad cerebrovascular o arterial periférica prematura (hombres < 55 años y mujeres < 60 años) | 1 |
| Examen físico | |
| Xantomas tendinosos | 6 |
| Arco corneal < 45 años | 4 |
| Análisis de laboratorio | |
| cLDL ≥ 330 mg/dl | 8 |
| cLDL 250-329 mg/dl | 5 |
| cLDL 190-249 mg/dl | 3 |
| cLDL 155-189 mg/dl | 1 |
| Análisis genético | |
| Mutación funcional en el gen del RLDL, APOB o PCSK9 | 8 |
| Diagnóstico de HF: | |
| Certeza: ≥ 8 puntos; probable: 6-7 puntos; posible: 3-5 puntos | |
De: WHO publication No. WHO7HGN/FH/CONS/99.213.
Los niveles plasmáticos elevados de triglicéridos no excluye el diagnóstico de HF cuando la historia familiar lo apoya. Los xantomas tendinosos son patognomónicos de HF; sin embargo, se encuentran en menos del 30% de los casos con diagnóstico genético de HF8,16, por lo que su ausencia no excluye el diagnóstico de HF. El diagnóstico diferencial de la HF se debe realizar con la hiperlipidemia familiar combinada, la hipercolesterolemia poligénica con agregación familiar y otras causas de hipercolesterolemia secundaria.
Diagnóstico en niños y adolescentesEl diagnóstico se puede sospechar en presencia de niveles de cLDL>190mg/dl o bien con cLDL>150mg/dl cuando se tiene la confirmación genética de HF o al menos la evidencia de transmisión vertical de la hipercolesterolemia y/o ECP en uno de los progenitores. Se ha demostrado que los niveles de cLDL discriminan bien entre aquellos niños con y sin HF antes de los 10años17. Para el diagnóstico, se recomienda obtener la media de 2 determinaciones del perfil lipídico con al menos 2 meses de diferencia debido a la variabilidad biológica en la edad infantil y descartar las causas de hipercolesterolemia secundaria en la infancia-adolescencia.
No hay un criterio único respecto a la edad en la que se debe hacer el diagnóstico de HF. En general se recomienda el diagnóstico entre los 2 y los 10años11,12,18-20. Su importancia es que cuanto antes se realice, más fácil será la adherencia a los hábitos de vida saludables. Este panel recomienda que el diagnóstico se debe realizar a partir de los 2años, especialmente cuando uno de los progenitores ya está diagnosticado, y a ser posible antes de los 8años.
Diagnóstico de la hipercolesterolemia familiar homocigotaLa hipercolesterolemia familiar homocigota (HFHo) es una forma rara de HF que se produce cuando se hereda la misma mutación en el gen del RLDL de ambos progenitores. Las mutaciones en los genes APOB, PCSK9 y la hipercolesterolemia autosómica recesiva (HAR) también pueden producir un fenotipo similar que varía en gravedad1,21. Se estima que afecta a un caso por millón de personas, aunque es mayor en determinadas regiones o países, presumiblemente debido a un efecto fundador y al aislamiento de una población.
El diagnóstico de HFHo se debe realizar alrededor de los 2años o inclusive antes y se basa en: concentración plasmática de cLDL sin tratamiento >500mg/dl o cLDL con tratamiento >300mg/dl, presencia de xantomas antes de los 10años (fig. 1) e historia de hipercolesterolemia o de diagnóstico genético en ambos progenitores21. Ambos padres deberían tener hipercolesterolemia y/o ser heterocigotos obligados para la misma mutación causante de HF.
Habitualmente se desarrolla una ateroesclerosis grave y generalizada que suele manifestarse clínicamente como enfermedad coronaria en edades muy jóvenes, así como con estenosis aórtica, y que si no se tratan pueden producir la muerte antes de los 20años21.
Diagnóstico genéticoLa principal ventaja de la detección de una variante funcional con efecto patogénico en los genes descritos anteriormente es que establece el diagnóstico inequívoco de la HF y facilita el cribado en cascada familiar22. En España se han descrito más de 400 mutaciones en el gen del RLDL asociadas a HF23. El diagnóstico genético solo se debe ofrecer a los CI con una puntuación ≥6 según los criterios de la RCLH, ya que tienen la mayor sensibilidad y especificidad15,24, se debe realizar en un laboratorio acreditado y debe incluir la secuenciación completa para identificar mutaciones puntuales y deleciones/inserciones para el gen del RLDL y de la APOB y PCSK9.
Detección de la hipercolesterolemia familiar: cribado en cascada familiarLa detección de la HF cumple los criterios de la Organización Mundial de la Salud (OMS) para el cribado sistemático de una enfermedad13 y es coste-efectiva para detectar nuevos casos de HF25–27. Una estrategia sistemática es esencial para la detección de los CI con HF. El CI es el primer miembro de una familia en ser diagnosticado y es fundamental para iniciar el cribado familiar en cascada. Los criterios diagnósticos de la RCLH (tabla 2) solo se deben utilizar para el diagnóstico del CI mayor de 18años y nunca en sus familiares13,15.
En la AP, se deben buscar los CI mediante la detección oportunista basada en la historia personal y/o familiar de hipercolesterolemia y ECP (antes de los 55años en varones y de los 60años en mujeres). A nivel hospitalario los CI se deben buscar entre los pacientes menores de 60años con enfermedad coronaria e hipercolesterolemia.
Para realizar el cribado en los familiares de primer grado de un CI diagnosticado de HF se recomienda usar una combinación de niveles de cLDL y análisis genético si se dispone de los recursos necesarios12,20,28–30. Diferentes estudios han demostrado que hasta el 24% de los familiares con un colesterol inferior al percentil 90 tienen un diagnóstico genético positivo, lo que justificaría realizar el análisis genético, ya que estas personas pueden transmitir el trastorno a su descendencia y expresar la hipercolesterolemia en edades más tardias14,27,31.
Con el fin de una mejor utilización de los recursos, se recomienda que el cribado sistemático en cascada sea coordinado por un servicio especializado y dedicado que colabore estrechamente con la AP e idealmente con una organización de pacientes12,19,27. Los pacientes deben ser informados de su RCV, de la importancia de informar a sus familiares para la detección precoz, así como de realizar el estudio genético si está disponible.
Riesgo cardiovascular en la hipercolesterolemia familiarLos pacientes con HF son considerados de alto RCV24,29. Sin embargo, el riesgo puede variar entre individuos en función de la presencia de otros factores de RCV (FRCV), especialmente la lipoproteína(a) [Lp(a)] y el tabaco, así como la presencia de ateroesclerosis subclínica24,32–34. La estratificación en niveles de riesgo ayuda al médico a individualizar la intensidad del tratamiento y permite una mejor utilización de los recursos.
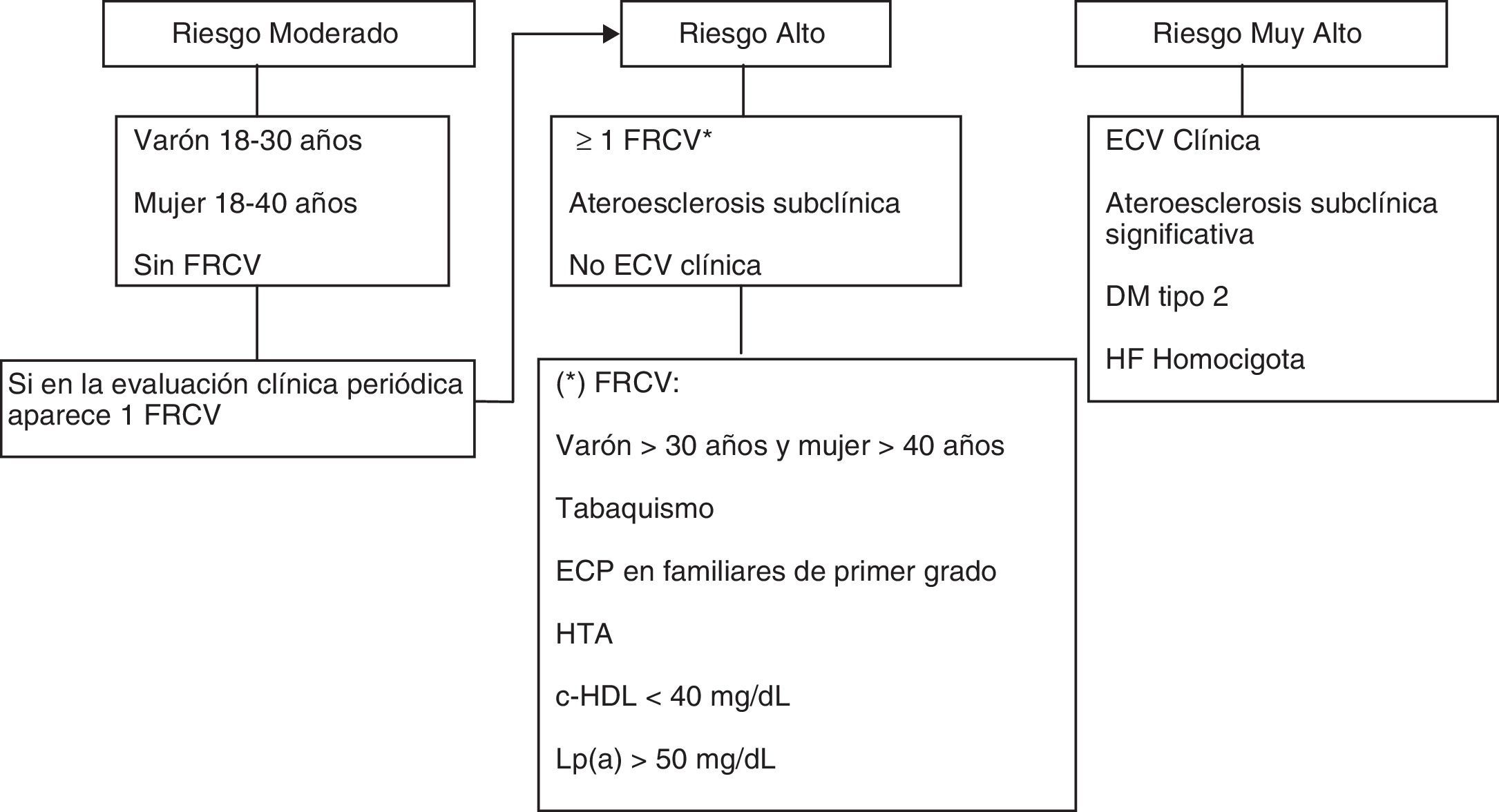
La estratificación del riesgo basada en Framingham o el SCORE no es adecuada en la HF, ya que lo infravaloran sistemáticamente, especialmente en los jóvenes11,12,24,29. En estos, una medida del RCV a largo tiempo basada en la imagen de ateroesclerosis subclínica podría ser adecuada.
La evaluación de la aterosclerosis coronaria subclínica se puede realizar de forma no invasiva, mediante la prueba de esfuerzo ECG, ecocardiografía de estrés, gammagrafía radioisotópica y el angio-TAC coronario32,35,36. La evaluación de otros territorios vasculares incluye la ecografía carotídea y la determinación del índice tobillo-brazo. Se recomienda evaluar la presencia de ateroesclerosis a partir de los 30años en varones y de los 40años en mujeres, o antes, si hay FRCV (fig. 2). En el caso de que alguna de las pruebas de imagen (angio-TAC y ecografía carotídea) muestre ateroesclerosis significativa (estenosis >50%) o una de las otras pruebas sea positiva, el paciente debe ser evaluado por Cardiología o Cirugía Vascular.
; ECV: enfermedad cardiovascular; FRCV: factores de riesgo cardiovascular; HF: hipercolesterolemia familiar; HTA: hipertensión arterial (elevación de la presión arterial sistólica ≥140mmHg, de la presión arterial diastólica ≥90mmHg, o ambas).")
Hipercolesterolemia familiar. Evaluación y estratificación del riesgo cardiovascular en adultos. c-HDL: colesterol transportado por lipoproteínas de alta densidad; DM: diabetes mellitus; ECP: enfermedad coronaria prematura (<55años en varones y <60años en mujeres); ECV: enfermedad cardiovascular; FRCV: factores de riesgo cardiovascular; HF: hipercolesterolemia familiar; HTA: hipertensión arterial (elevación de la presión arterial sistólica ≥140mmHg, de la presión arterial diastólica ≥90mmHg, o ambas).
Las Guías Internacionales en HF del 2004 estratificaron el RCV en alto, intermedio y bajo32. La presencia de un grupo de bajo riesgo dentro de un trastorno que globalmente se considera de alto riesgo puede inducir a confusión. Por tanto, este panel de expertos recomienda clasificar aquellos pacientes con HFHo, HF con enfermedad coronaria o con diabetes mellitus tipo2 (DM2), o aquellos con evidencia de enfermedad ateroesclerótica subclínica significativa como de muy alto RCV29. Los pacientes con al menos un FRCV deben considerarse de alto RCV y el resto de pacientes, especialmente los jóvenes, se podrían considerar de RCV moderado (fig. 2).
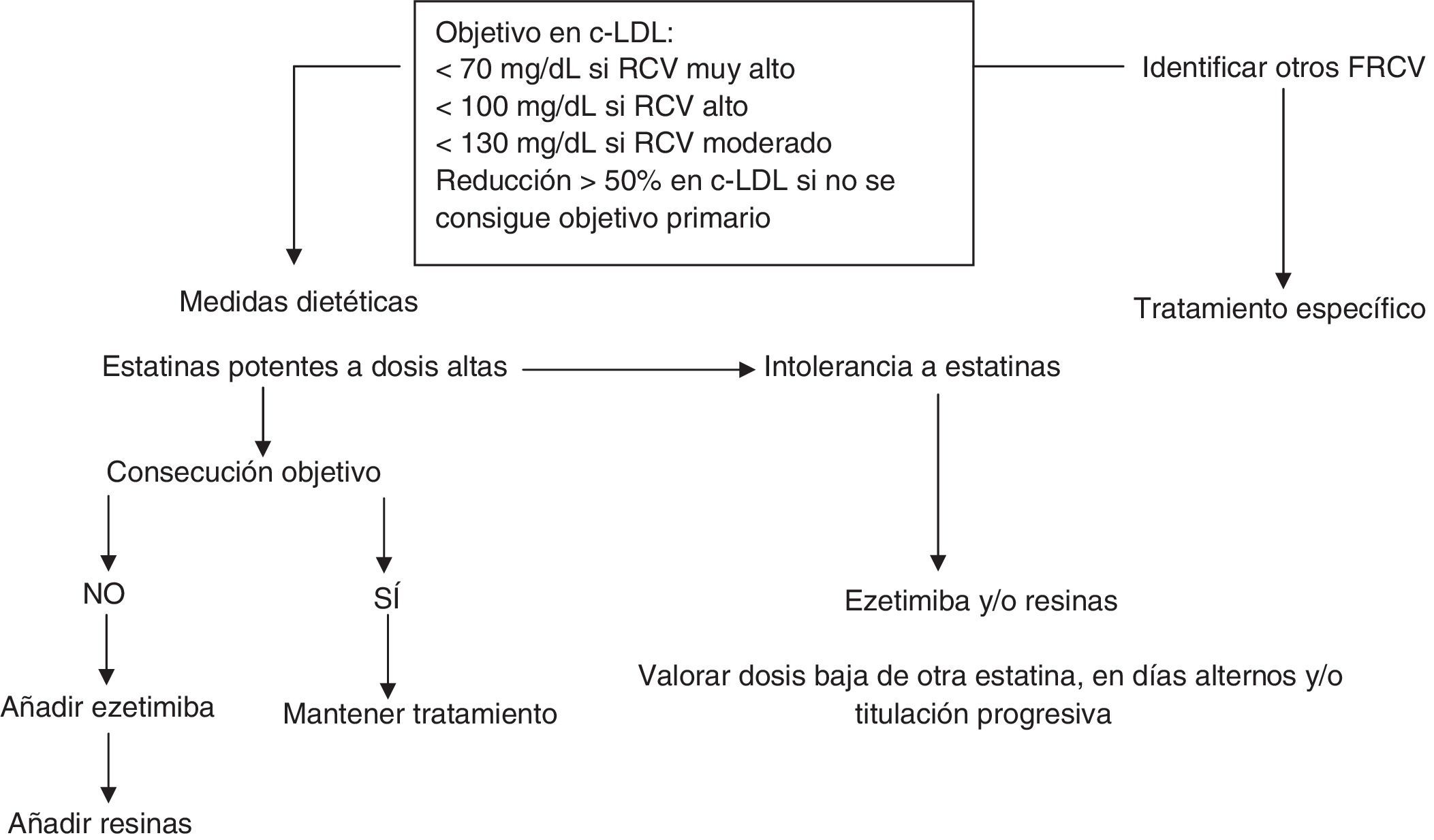
Objetivo terapéutico y tratamiento en adultosEste documento recomienda un objetivo en cLDL plasmático <100mg/dl en adultos con al menos un FRCV y <70mg/dl si existe enfermedad coronaria, DM2 o la presencia de enfermedad ateroesclerótica subclínica significativa. En el resto de los pacientes (varones <30años y mujeres <40años) que no tienen otro FRCV, un cLDL <130mg/dl se podría considerar aceptable (fig. 3). Sin embargo, debido a la dificultad de conseguir este objetivo en la mayoría de los pacientes7,8, una reducción del cLDL≥50% puede considerarse como un objetivo secundario más realista.

Todo adulto con HF debe ser tratado con medidas dietéticas y con fármacos hipolipemiantes desde el momento del diagnóstico. Varios estudios han demostrado que el tratamiento hipolipemiante intensivo tiene efectos beneficiosos en la reducción del grosor íntima-media carotídeo y en la mejora de la función endotelial37,38. Además, estudios observacionales han confirmado el beneficio cardiovascular de las estatinas en los pacientes con HF6,39. La base del tratamiento es el uso de una estatina potente como atorvastatina o rosuvastatina en monoterapia o en combinación con ezetimiba y/o resinas si no se consigue el objetivo terapéutico.
Recomendamos que la mayoría de estos pacientes sean seguidos en AP, y los casos complejos, en centros especializados.
Objetivo terapéutico y tratamiento en niños y adolescentesExiste acuerdo en que los objetivos de tratamiento en cLDL en los niños no necesitan ser tan bajos como en los adultos y no existe evidencia para un objetivo absoluto o relativo18,19. Este panel recomienda un cLDL plasmático <130mg/dl a partir de los 14años y <160mg/dl en los menores de 14años, excepto si hay otro FRCV o antecedentes de enfermedad coronaria muy prematura en el progenitor afecto, en que los objetivos pueden ser más estrictos.
La alimentación es la base del tratamiento de la HF en niños y adolescentes, consiguiéndose reducciones en cLDL de hasta un 15%20,40. Es esencial un aporte adecuado de energía y nutrientes para mantener un adecuado crecimiento y peso corporal. Además de una correcta alimentación, debe promoverse la actividad física así como no fumar.
No existe unanimidad sobre a qué edad se debe comenzar el tratamiento con estatinas en niños con HF y no hay datos de seguridad antes de los 8años11,12. En los heterocigotos, se recomienda el uso de estatinas a partir de los 10años en niños y preferiblemente después de la menarquia en las niñas18–20, y en los homocigotos la medicación debe ser iniciada en el momento del diagnóstico18. Las estatinas son seguras y eficaces en la población infantil41. Se puede utilizar cualquier estatina aprobada por las agencias reguladoras titulando la dosis según la respuesta clínica y la edad. Si con las estatinas no se consiguen los objetivos en cLDL, se debe valorar el añadir resinas o ezetimiba. Las estatinas están contraindicadas en el embarazo, por lo que debe advertirse a las adolescentes.
LDL-aféresisEs el tratamiento de elección para los pacientes con HFHo y ha demostrado tener un efecto beneficioso en la ateroesclerosis aórtica y coronaria41, mejorando la supervivencia. Permite la eliminación específica de cLDL y Lp(a) con una disminución plasmática del 50-75% cuando se usa de forma semanal o cada 2semanas42. El tratamiento con estatinas se debe mantener para retrasar el efecto rebote en el aumento del cLDL.
Con la evidencia disponible se puede recomendar su indicación en las siguientes situaciónes42,43: a)HFHo a partir de los 6años y siempre antes de los 10años; b)HF con enfermedad coronaria sintomática y cLDL>200mg/dl a pesar de tratamiento farmacológico intenso, y c)HF con enfermedad coronaria progresiva sin posibilidades de revascularización y cLDL>125mg/dl y Lp(a)>60mg/dl a pesar de tratamiento farmacológico intenso.
Papel de las organizaciones de pacientes en la detección de la hipercolesterolemia familiar y en el apoyo familiarSe estima que en España hay unas 100.000 personas con HF, y la esperanza de vida de estos pacientes puede verse reducida de 20 a 30años, lo que se traduciría en la pérdida potencial de 2 millones de años de vida. Además, el paciente con HF tiene una buena calidad de vida, que empeora con la presencia de enfermedad coronaria44.
Una organización de pacientes con HF desempeña una importante función en el apoyo a los pacientes y sus familias, promoviendo el conocimiento de la HF en la comunidad, los médicos y sistemas de salud. Además, una organización de pacientes puede proporcionar los medios para establecer un registro tanto de pacientes como de los servicios disponibles, incluyendo los centros que realizan el cribado en cascada para la detección de la HF.
En 1997, y de acuerdo con las recomendaciones de la OMS, se crea en España la Fundación de Hipercolesterolemia Familiar (FHF), organización benéfica asistencial sin ánimo de lucro (www.colesterolfamiliar.org). Su misión es informar, educar, detectar y apoyar a las familias con HF. Entre los logros importantes destaca la obtención de la aportación reducida al tratamiento crónico con estatinas y ezetimiba.
En los últimos años, algunas comunidades autónomas están realizando el diagnóstico genético de HF financiado por el sistema de salud, y en la actualidad hay más de 6.000 pacientes en España identificados genéticamente, cerca del 50% gracias al programa de detección de la FHF en colaboración con centros hospitalarios de toda España (http://safeheart.colesterolfamiliar.org/).
Programas de detección regionalesEn España, las comunidades de Aragón, Castilla y León, Cataluña, La Rioja, Madrid, Navarra y País Vasco han implementado diferentes estrategias de detección de la HF, incluyendo el diagnóstico genético.
Castilla y León ha sido la única comunidad que en su estrategia de detección ha incluido a los médicos de AP (http://www.saludcastillayleon.es). Esta comunidad, en colaboración con la FHF, viene desarrollando desde 2009 el Programa de detección precoz de la HF, que incluye la formación de los médicos y la creación de un registro ad hoc. El médico de AP o de atención especializada selecciona el CI siguiendo los criterios diagnósticos de la RCLH (puntuación ≥6). Una vez identificado, se solicita el estudio genético en muestra de saliva. Si se confirma el diagnóstico se procede a realizar la detección en cascada familiar a los familiares de primer grado. Hasta la fecha se han diagnosticado genéticamente unos 1.000 pacientes con HF. Este programa debería servir de modelo para el resto de comunidades que todavía no lo están realizando y para impulsar una estrategia homogénea a nivel nacional.
Resumen de recomendaciones1. Diagnóstico de hipercolesterolemia familiar en adultos1.1. Se debe sospechar una HF en un adulto con cLDL>220mg/dl y antecedentes de hipercolesterolemia y/o ECP en familiares de primer grado. (A)
1.2. Los criterios de la RCLH se deben utilizar solamente para el diagnóstico del caso índice (CI) y siempre en >18 años. (A)
1.3. Para detectar a los CI se debe realizar una búsqueda oportunista en los pacientes con ECP y en aquellos con antecedentes personales y/o familiares de hipercolesterolemia y/o ECP. (A)
2. Diagnóstico de hipercolesterolemia familiar en niños y adolescentes2.1. Se debe sospechar una HF en niños con cLDL≥190mg/dl o cLDL>150mg/dl con historia de hipercolesterolemia y/o ECP en uno de los progenitores. (A)
2.2. Se debería realizar la determinación de colesterol en los niños a partir de los 2años si existe historia de HF en uno de los progenitores. (A)
2.3. Si se conoce el defecto genético en uno de los padres, se puede realizar el diagnóstico genético en el niño con cLDL>150mg/dl, previa información y consentimiento del progenitor afectado o tutor legal. (A)
3. Diagnóstico de hipercolesterolemia familiar homocigota3.1. Se debe sospechar en presencia de un cLDL>500mg/dl sin tratamiento. (A)
3.2. Se debe sospechar en presencia de un cLDL>300mg/dl con el máximo tratamiento farmacológico. (B)
3.3. La presencia de xantomas tuberosos o tendinosos antes de los 10años de edad sugiere el diagnóstico de HFHo. (A)
4. Diagnóstico genético y cribado familiar4.1. Se recomienda realizar el diagnóstico genético de HF en un CI con una puntuación ≥6 puntos de acuerdo a criterios clínicos de la RCLH. (A)
4.2. La no detección de una mutación no excluye el diagnóstico de HF cuando el fenotipo es sugerente. (B)
4.3. El cribado en cascada familiar debería combinar niveles de colesterol total y cLDL y análisis genético si está disponible. (A)
4.4. El cribado en cascada familiar debería estar centralizado y requiere la coordinación entre los médicos de AP y de atención especializada así como de enfermería. (A)
5. Riesgo cardiovascular en la hipercolesterolemia familiar5.1. Los pacientes con HF se deben considerar de alto RCV. Sin embargo, en algunos casos se podría estratificar el riesgo. (A)
5.2. Los pacientes con HF y con al menos un FRCV tienen un RCV alto. (A)
5.3. Los pacientes con HFHo y con HF que presentan ateroesclerosis subclínica significativa, enfermedad coronaria clínica o DM2 tienen un RCV muy alto. (A)
5.4. Se deben medir los niveles de Lp(a) en todos los pacientes con HF. (A)
5.5. En pacientes con HF heterocigota >30años (varones) y >40años (mujeres) sin clínica de enfermedad coronaria se debería realizar una búsqueda de enfermedad aterosclerótica mediante técnicas no invasivas, aunque su utilidad no se ha validado en la HF. (C)
6. Objetivo y tratamiento de hipercolesterolemia familiar en adultos6.1. En los pacientes con HF y RCV muy alto, el objetivo de cLDL debería ser <70mg/dl. (A)
6.2. En los pacientes con HF y RCV alto, el objetivo de cLDL debería ser <100mg/dl. (A)
6.3. En el resto de los pacientes adultos con HF se podría considerar un cLDL <130mg/dl. (B)
6.4. En el caso de no alcanzar el objetivo en cLDL con el máximo tratamiento tolerado, se debe conseguir al menos una reducción de cLDL≥50%. (A)
6.5. Se recomienda la administración de estatinas potentes a dosis altas y, si es necesario, en combinación con ezetimiba y/o resinas. (A)
7. Objetivo y tratamiento de hipercolesterolemia familiar en niños y adolescentes7.1. Se puede comenzar tratamiento con estatinas a partir de los 10años en los varones y preferiblemente después de la menarquia en las niñas. (A)
7.2. El objetivo en cLDL debería ser <130mg/dl y se podría considerar un cLDL<160mg/dl en los <14 años sin otro FRCV. (B)
7.3. Es necesario monitorizar el crecimiento y el desarrollo puberal, así como los niveles de transaminasas y creatincinasa al inicio, a los 3meses de tratamiento y posteriormente de forma anual. (A)
7.4. Los niños con HFHo deben iniciar el tratamiento farmacológico en el momento del diagnóstico e idealmente no más tarde de los 2años. (A)
8. LDL-aféresis8.1. La LDL-aféresis se puede realizar en los pacientes con HFHo a partir de los 6años y en casos seleccionados de HF heterocigota grave. (A)
8.2. El efecto de la LDL-aféresis en la progresión de la ateroesclerosis se debe monitorizar con ecocardiografía de la válvula y raíz aórtica, ecografía de carótida y test de esfuerzo. (B)
FinanciaciónEste trabajo ha sido apoyado por la Fundación Hipercolesterolemia Familiar. El programa de detección genética de la HF en cascada familiar ha contado con fondos de la Red de Hiperlipemias Genéticas del Instituto de Salud CarlosIII (ISCIII) G03/181, Centro Nacional de Investigación Cardiovascular (CNIC) 08-2008 y FIS PI12/01289 del ISCIII.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Agradecemos al Dr. Gerald Watts (Universidad de Perth, Australia) por la lectura del documento y por sus constructivos y expertos consejos. A los Drs. Rosa de los Ríos y Guillermo Domenech, de la Dirección General de Salud Pública de la consejería de Castilla y León, por su contribución al programa de detección de la HF en esa Comunidad. A María Teresa Pariente, de la FHF, por su dedicación en la coordinación del programa de detección en cascada familiar de la HF, y a las familias por su participación.
Coordinador:
Pedro Mata.
Colaboradores del Documento de Consenso:
Feliciano J. Ramos (Hospital Clínico Universitario Lozano Blesa, Zaragoza).
Catalina Sánchez (Fundación Hipercolesterolemia Familiar, Madrid).
Gerardo Gonzalo (Fundación Hipercolesterolemia Familiar, Madrid).
José Javier Castrodeza (Ex Director de Salud Pública, Consejería de Sanidad, Castilla y León).
José L. Zamorano (Hospital Ramón y Cajal, Madrid).
Asesor Internacional:
Gerald F. Watts (Lipid Disorders Clinic, Department of Internal Medicine, Royal Perth Hospital, University of Western Australia, Australia).
Organización Promotora:
Fundación Hipercolesterolemia Familiar.
recomendados

Medicina de Familia. SEMERGEN sigue las recomendaciones para la preparación, presentación y publicación de trabajos académicos en revistas biomédicas