















Los médicos de familia estamos acostumbrados a diagnosticar y tratar enfermedades pulmonares crónicas como la EPOC y el asma debido a su alta prevalencia en nuestras consultas, pero es importante realizar un diagnóstico diferencial ante pacientes con disnea que tras descartar estas 2 enfermedades obstructivas pueden presentar otras enfermedades respiratorias menos frecuentes o raras.
Según la definición de la Unión Europea, las enfermedades raras, minoritarias o huérfanas son las enfermedades con peligro de muerte o de invalidez crónica que tienen una prevalencia menor de 5 casos por cada 10.000 habitantes. Dentro de las enfermedades respiratorias, la fibrosis pulmonar idiopática (FPI) cumple estos criterios y tiene una prevalencia estimada en España entre 13 casos por cada 100.000 habitantes en mujeres y 20 por cada 100.000 en varones. Aunque su historia natural puede ser variable e impredecible, en general la supervivencia media es de 2-5 años desde el inicio de los síntomas1,2, por lo que su sospecha clínica y diagnóstico precoz resultan especialmente relevantes.
La FPI es la enfermedad pulmonar intersticial difusa (EPID) más frecuente seguida de la sarcoidosis. Las EPID constituyen unos grupos de afecciones pulmonares con manifestaciones clínicas, radiológicas y funcionales respiratorias similares, en las que las principales alteraciones anatomopatológicas afectan las estructuras alveolointersticiales, aunque en muchas ocasiones también afectan las pequeñas vías respiratorias, así como la vasculatura pulmonar.
Su clasificación ha sido revisada recientemente. Se dividen en 3 grupos3 (tabla 1):
Clasificación EPID
| Neumonías intersticiales idiopáticas |
| Neumonías intersticiales idiopáticas |
| Fibrosis pulmonar idiopática |
| Neumonía intersticial aguda |
| Neumonía intersticial no específica idiopática |
| Bronquiolitis respiratoria con enfermedad pulmonar intersticial |
| Neumonía intersticial descamativa |
| Neumonía organizada criptogenética |
| Neumonías intersticiales idiopáticas raras |
| Neumonía intersticial linfocítica idiopática |
| Fibroelastosis pleuropulmonar idiopática |
| Neumonías intersticiales idiopáticas no clasificables |
| De causa conocida o asociadas |
| Asociadas a enfermedades del colágeno |
| Causadas por polvos inorgánicos (neumoconiosis) |
| Inducidas por fármacos y radioterapia |
| Causadas por polvos orgánicos (neumonitis por hipersensibilidad) |
| Asociadas a enfermedades hereditarias |
| Primarias o asociadas a otros procesos no bien definidos |
| Sarcoidosis |
| Proteinosis alveolar |
| Microlitiasis alveolar |
| Linfangioleiomiomatosis |
| Eosinofilias pulmonares |
| Histiocitosis X (granulomatosis de células de Langerhans) |
| Amiloidosis |
Neumonías intersticiales idiopáticas: constituido por entidades clínicopatológicas cuya definición histológica ha suscitado gran atención en los últimos años.
De causa conocida o asociadas: en este grupo se incluyen las manifestaciones pulmonares de las enfermedades del colágeno, que con frecuencia tienen una histología indistinguible de las neumonías intersticiales idiopáticas. En este grupo también se incluyen las EPID producidas por fármacos, polvos orgánicos (alveolitis alérgicas extrínsecas, también denominadas neumonitis por hipersensibilidad), polvos inorgánicos (neumoconiosis) y las asociadas a enfermedades hereditarias.
Primarias o asociadas a otros procesos no bien definidos: formado por un conjunto de entidades idiopáticas pero con clínica e histología bien definidas.
Este monográfico se centrará exclusivamente en la FPI, intentando que desde atención primaria seamos capaces de reconocer ese pequeño grupo de pacientes que la padecen, ya que su diagnóstico precoz puede ayudar a iniciar el tratamiento de forma temprana, con el fin de que su calidad de vida sea la mejor posible. Una vez identificados los pacientes que presentan los síntomas y signos típicos de la FPI deben ser derivados a un servicio de neumología con experiencia en esta enfermedad para confirmar el diagnóstico y optimizar su manejo.
Fibrosis pulmonar idiopáticaLa FPI se define como una enfermedad de etiología desconocida, limitada a los pulmones, que cursa con fibrosis progresiva y se caracteriza por una fisiología pulmonar restrictiva y alteraciones radiológicas e histológicas de neumonía intersticial usual (NIU).
Se manifiesta habitualmente más allá de la quinta década de la vida y afecta con mayor frecuencia a varones. Su diagnóstico requiere la exclusión de otras formas de neumonía intersticial tanto idiopáticas como asociadas a enfermedades sistémicas, exposicionales o por fármacos1.
EpidemiologíaEn cuanto a su incidencia se estima que varía entre 4,6-7,4/100.000 habitantes, diagnosticándose en la Comunidad Europea entre 30.000 y 40.000 casos al año. Su prevalencia, comentada anteriormente, se sitúa entre 13/100.000 habitantes en mujeres y 20/100.000 habitantes varones. De acuerdo a estos datos se estima que en España la FPI puede estar afectando a unas 7.000-12.000 personas y que afecta a más de 5 millones de personas en el mundo. Como se ha comentado en la introducción se trata de una enfermedad rara, y como tal destaca su alta mortalidad, que recordemos no supera los 2-5 años de supervivencia media tras el inicio de los síntomas2.
Historia naturalSu historia natural es variable e impredecible en el momento del diagnóstico (fig. 1). Algunos pacientes pueden permanecer asintomáticos durante 2-3 años. La mayoría desarrollan una lenta progresión, con deterioro clínico y funcional respiratorio que finalmente ocasiona insuficiencia respiratoria crónica. Otros casos presentan periodos de relativa estabilidad con episodios de agudización (exacerbaciones agudas u otras complicaciones) que son causa de una elevada morbimortalidad. En una minoría de los pacientes la enfermedad es de corta duración, con una progresión más rápida (forma acelerada)1,2,4.

Aunque se considera idiopática, se cree que su desarrollo probablemente es debido al efecto de diversos factores ambientales en sujetos con predisposición genética. Las alteraciones genéticas con más relevancia clínica son las mutaciones en los genes que mantienen la longitud de los telómeros (TERT, TERC), que son más frecuentes en las formas familiares, en la proteína C del surfactante y en la región promotora de la mucina 5B (MUC5B), este último se ha relacionado recientemente con los pacientes de progresión más rápida.
Junto al tabaquismo (hay estudios que encuentran una mayor probabilidad de desarrollar FPI en pacientes con historia de tabaquismo de 20 paquetes/año)5 se consideran factores de riesgo para la FPI la exposición al sílice, al latón, al acero, al plomo y al polvo de la madera,asi como las actividades laborales de la ganadería y agricultura. Hay estudios que consideran al reflujo gastroesofágico (RGE) como factor de riesgo para la predisposición y la progresión de la FPI2.
DiagnósticoManifestaciones clínicasSíntomasLa FPI se presenta con mayor frecuencia entre la quinta y séptima décadas de la vida. Dos tercios de los casos se presentan en mayores de 60 años. Los síntomas más frecuentes son la disnea de esfuerzo y la tos.
La disnea de esfuerzo es el síntoma cardinal. Es lentamente progresiva y durante un tiempo puede ser el único síntoma, por lo que los pacientes acuden al médico meses después del inicio de la enfermedad.
La tos generalmente es seca, improductiva. En algunos pacientes asintomáticos, la enfermedad se sospecha ante las alteraciones observadas en la radiografía de tórax. Estudios retrospectivos sugieren que los síntomas pueden preceder al diagnóstico entre 6 meses y 2 años6.
Exploración físicaAuscultación respiratoria. Es característica la presencia de estertores crepitantes desde fases tempranas. Los crepitantes finos inspiratorios bibasales (crackles) pueden orientarnos al diagnóstico de FPI. Se auscultan predominantemente en las bases y campos posteriores pulmonares, tienen un sonido que recuerda al producido cuando se separa cuidadosamente una cinta de velcro7 y se detectan mejor durante una respiración lenta y profunda. Estos estertores crepitantes se auscultan en el 90% de los pacientes2,8 (fig. 2).

Exploración general. Las acropaquias (dedos en palillos de tambor) son frecuentes y se objetivan en el 50% de los casos2,7 (fig. 3). No hay otras manifestaciones propias de la enfermedad, aunque pueden estar presentes las debidas a la asociación de comorbilidades como hipertensión pulmonar, enfisema, síndrome de apneas hipoapneas del sueño, cáncer de pulmón o RGE1,2. En las etapas más avanzadas de la enfermedad se presentan los síntomas y signos de insuficiencia cardiaca derecha propios del cor pulmonale (éstasis yugular, hepatomegalia, edemas).
Sospecha diagnóstica
Teniendo en cuenta que la demora en el diagnóstico de la enfermedad puede llegar a los 2 años, y aunque la enfermedad es poco frecuente, por su elevada morbimortalidad y mal pronóstico es importante el diagnóstico precoz, motivo por el que el médico de familia juega un papel esencial en sospecha diagnóstica. La sospecha diagnóstica de FPI debe plantarse desde atención primaria ante pacientes mayores de 50 años que consultan por disnea de esfuerzo de curso progresivo y tos seca.
La disnea es un motivo de consulta frecuente en atención primaria y el médico de familia suele ser quien primero valora la causa de esa disnea. Es un síntoma de alerta al que el médico debe prestar una atención especial y realizar un diagnóstico diferencial completo, pues puede ser la manifestación inicial de una enfermedad de base cardiológica, neumológica, metabólica, muscular, neoplásica o incluso psicógena. Realizar una anamnesis minuciosa es básico para la adecuada orientación diagnóstica. Para la valoración de la disnea se utilizan escalas de fácil manejo, como la escala de disnea modificada del Medical Research Council (MRC)9.
El origen de la disnea puede ser más difícil de determinar cuando se desarrolla progresivamente a lo largo de meses. En la mayoría de los pacientes con disnea de causa no aclarada, una de las siguientes 4 enfermedades suele ser la causa: asma, EPOC, enfermedad pulmonar intersticial o miocardiopatía10. Sin embargo, en la práctica clínica la causa de la disnea persistente en un paciente puede atribuirse comúnmente al envejecimiento, a una enfermedad cardiaca, o a la EPOC, lo que se traduce en retrasos en el diagnóstico.
La historia clínica detallada y una exploración física completa (recordar el papel esencial de la auscultación) pueden orientar definitivamente en la identificación de la causa de la disnea y descartar la causa cardiaca, enfermedad sistémica o enfermedades pulmonares obstructivas. Desechadas estas, ante un paciente con disnea y tos debe realizarse un enfoque diagnóstico orientado a la posible presencia de una EPID, teniendo en cuenta también que la EPID más frecuente es la FPI, seguida de la sarcoidosis, las neumonitis por hipersensibilidad y las EPID asociadas a las enfermedades del colágeno11. En un paciente mayor de 50 años con disnea de esfuerzo de curso progresivo, tos seca y estertores crepitantes basales inspiratorios debemos pensar en FPI.
Ante la posible presencia de EPID en la tercera parte de los casos se puede elaborar un diagnóstico de sospecha con una anamnesis adecuada12. Debe tenerse en cuenta que el diagnóstico de FPI es un diagnóstico de exclusión. La anamnesis, además de recoger las características de la disnea y de la tos, tiene que dirigirse especialmente hacia los siguientes puntos1,2:
- -
Edad: la FPI suele diagnosticarse en sujetos mayores de 50 años.
- -
Antecedentes familiares: los pacientes con FPI pueden tener algún otro miembro de la familia afectado (FP familiar).
- -
Hábito tabáquico: el tabaquismo se asocia con FPI.
- -
Historia ocupacional: se deben registrar las actividades realizadas por el paciente en su vida profesional, así como los ambientes laborales a los que ha estado expuesto.
- -
Fármacos: los fármacos son causa de EPID por lo que se deben revisar los fármacos que toma o ha tomado el paciente, dosis y duración del tratamiento.
- -
Enfermedades sistémicas: hay que valorar la presencia de síntomas o signos de enfermedades sistémicas como colagenosis o sarcoidosis que pueden asociarse a EPID o a neumopatías intersticiales idiopáticas.
- -
Radioterapia: la exposición a radioterapia puede ser causa de EPID.
Las pruebas complementarias a utilizar en asistencia primaria ante un paciente con disnea y sospecha de EPID son la radiografía de tórax y la espirometría, además del electrocardiograma, para descartar la causa cardiaca de la sintomatología y análisis sanguíneos generales en el contexto de una valoración general.
Radiografía de tóraxLa radiografía de tórax en proyección póstero-anterior y lateral es la exploración complementaria inicial imprescindible en un paciente con disnea. Es una prueba básica en la evaluación inicial en un paciente en el que se sospeche una EPID o FPI. La mayoría de las radiografías de tórax son anormales. El 90% de los pacientes con FPI presentan alteraciones radiográficas en el momento del diagnóstico. El patrón radiológico de la FPI consiste en imágenes reticulares, bilaterales, simétricas, con predilección en los lóbulos inferiores. En fases avanzadas puede observarse el patrón en panal de abeja (fig. 4).

No obstante, la radiografía de tórax carece de la precisión diagnóstica adecuada, de forma que un diagnóstico correcto de la FPI se realiza por radiografía de tórax en menos del 50% de los casos. Por otra parte, la interpretación de patrones intersticiales tiene una baja concordancia entre distintos observadores7.
Espirometría forzadaConstituye una prueba básica y al alcance en la actualidad de los médicos de atención primaria, y bien realizada, es una herramienta de ayuda en la orientación diagnóstica de un paciente en el que se sospeche FPI.
En la espirometría forzada, el patrón funcional se caracteriza por un trastorno restrictivo (especialmente debido a la disminución de la capacidad vital forzada [FVC]). La relación FEV 1/FVC permanece aumentada o normal. No obstante, una espirometría normal no excluye el diagnóstico de FPI. La presencia de un patrón obstructivo descarta en principio la FPI.
Derivación de los pacientes a servicios de neumologíaAnte un paciente con cuadro clínico sugerente de FPI (tabla 2) hay que derivar para estudio y confirmación a un servicio de neumología, con experiencia en el diagnóstico de EPID. Un diagnóstico precoz es clave para el inicio del tratamiento. Es aceptado que los pacientes deben ser derivados a centros con experiencia en EPID para confirmar el diagnóstico y ofrecer opciones terapéuticas13.
Sospecha de FPI
| Comienzo insidioso |
| Edad entre 40-80 años |
| Tos no productiva |
| Disnea de esfuerzo lentamente progresiva |
| Crepitantes secos, bibasales, inspiratorios, tipo «velcro» |
| Acropaquias o dedos en palillo de tambor |
| Pruebas funcionales respiratorias anormales, de predominio restrictivo y con deterioro del intercambio de gases |
Con estos criterios se recomienda estudio radiológico y derivación a servicios de neumología para estudio
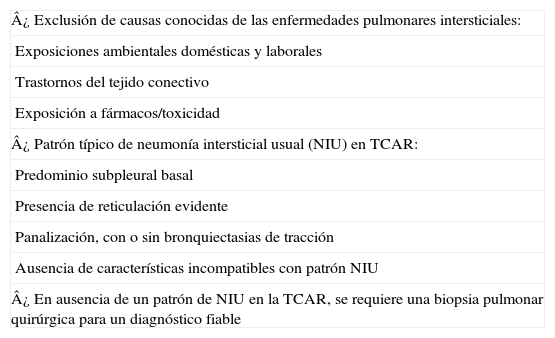
Actualmente se recomienda basar el diagnóstico en el trípode clínico, radiológico y patológico mediante un equipo multidisciplinar en el que participen neumólogos, radiólogos y patólogos expertos en el diagnóstico y manejo de las EPID pues se consigue aumentar la precisión diagnóstica1,2,14. Para la confirmación diagnóstica definitiva de FPI se requiere:a) La exclusión de otras entidades clínicas definidas o enfermedades parenquimatosas pulmonares difusas de causa conocida (exposición ambiental u ocupacional, enfermedades del tejido conectivo, toxicidad por fármacos), y b) la presencia de un patrón histológico de NIU en el examen del tejido pulmonar obtenido mediante biopsia pulmonar quirúrgica, o bien la evidencia radiológica de patrón NIU en la tomografía axial computarizada de alta resolución (TACAR), o ambas1,2 (tabla 3) (fig. 5).
Criterios diagnósticos de FPI
| ¿ Exclusión de causas conocidas de las enfermedades pulmonares intersticiales: |
| Exposiciones ambientales domésticas y laborales |
| Trastornos del tejido conectivo |
| Exposición a fármacos/toxicidad |
| ¿ Patrón típico de neumonía intersticial usual (NIU) en TCAR: |
| Predominio subpleural basal |
| Presencia de reticulación evidente |
| Panalización, con o sin bronquiectasias de tracción |
| Ausencia de características incompatibles con patrón NIU |
| ¿ En ausencia de un patrón de NIU en la TCAR, se requiere una biopsia pulmonar quirúrgica para un diagnóstico fiable |

Algoritmo diagnóstico de la FPI2.
Es la prueba fundamental para el diagnóstico de la FPI al identificar los hallazgos típicos del patrón de NIU establecidos por el consenso oficial ATS/ERS/JRS/ALAT 20111 (tabla 3) (fig. 6).
.")
Se recomienda la lectura radiológica utilizando una terminología descriptiva basada en la correlación radiológico-patológica, como la aconsejada por la sociedad Fleischner15. La tomografía axial computarizada de alta resolución (TACAR) tiene un valor predictivo positivo en el diagnóstico de la NIU del 90-100%. Además puede mostrar la presencia de comorbilidades como el enfisema, la hipertensión pulmonar o el cáncer de pulmón.
Biopsia pulmonar quirúrgicaEs la prueba que da el diagnóstico definitivo cuando en la TACAR no se observa un patrón de certeza típico de NIU. El patrón histológico de NIU viene recogido en la figura 7.

Otras pruebas diagnósticas que son de utilidad en el manejo diagnóstico y el seguimiento de la FPI están reflejadas en la tabla 4.
Pruebas de utilidad en el diagnóstico y seguimiento de la FPI
| ¿ Grado disnea (MRC, Escala Borg, Índice de disena basal) |
| ¿ Grado tos (Cuestionario de Leicester) |
| ¿ TCAR |
| ¿ Pruebas funcionales respiratorias (PFR): espirometría, pletismografía, DLco |
| ¿ Prueba de la marcha de los 6 minutos (PM6M) |
| ¿ Oximetría/gasometría arterial |
| ¿ Ecocardiograma (si sospecha de HTP) |
| ¿ Cuestionarios calidad de vida (SF-36, St George, UCSD) (opcional) |
UCDS: University of California San Diego Shortness of Breath Questionnaire.
Pletismografía. La medición de los volúmenes pulmonares estáticos confirma la fisiología restrictiva que se observa en la espirometría forzada. La alteración de los volúmenes por lo general se manifiesta en la reducción de la capacidad pulmonar total (TLC).
Capacidad de difusión (DLco). El intercambio de gases se deteriora en la FPI, y puede demostrarse por la medición de la DLco. Su disminución a veces puede preceder a los cambios en los volúmenes pulmonares y puede constituir la única alteración funcional durante las primeras etapas de la FPI.
La gasometría arterial en reposo suele ser normal. Hipoxemia y alcalosis respiratoria leve pueden presentarse en las fases avanzadas de la enfermedad, aunque en reposo la saturación arterial de oxígeno suele ser normal, es común la desaturación durante el ejercicio.
Pruebas de laboratorioNo hay alteraciones de laboratorio específicas para la FPI. A pesar de ello y sin clínica de enfermedad sistémica se deben realizar pruebas serológicas de autoinmunidad, observándose pruebas positivas hasta en un 20% de los casos1,8. En la actualidad se investiga la utilidad de biomarcadores para el diagnóstico y caracterización de la FPI16.
Lavado broncoalveolarEl lavado broncoalveolar es de gran utilidad en el estudio de la EPID. En la FPI se utiliza para el diagnóstico diferencial de otras entidades, en especial la neumonitis por hipersensibilidad1,2.
Biopsia transbronquialA diferencia de otras EPID como la sarcoidosis, la biopsia transbronquial no tiene utilidad en el diagnóstico de la FPI, dado que no es posible observar la distribución de la lesión por el tamaño de la muestra1,2.
TratamientoLas opciones de tratamiento precisan del conocimiento del grado de evolución de la enfermedad, su pronóstico y de las comorbilidades que se puedan asociar. Esto es fundamental en cuanto que determinadas opciones terapéuticas solo son válidas en función del estadio de gravedad y del estado evolutivo y pronóstico. Las bases para el tratamiento son1,2:
- -
La acción terapéutica primordial es la elección del tratamiento antifibrótico.
- -
Tratamiento sintomatológico de la tos y la disnea.
- -
Tratamiento de las comorbilidades: RGE, infecciones respiratorias, hipertensión pulmonar, síndrome de apneas-hipoapneas del sueño.
- -
Tratamiento no farmacológico: oxigenoterapia, rehabilitación respiratoria.
- -
En los estadios evolutivos avanzados, deberá considerarse la opción del trasplante pulmonar.
- -
Cuidados paliativos adecuada para asistir al paciente en la fase final de la enfermedad.
El conocimiento de las condiciones fisiopatológicas que atribuyen una preeminencia de base fibrótica en detrimento de un concepto previo de base inflamatoria, ha condicionado una nueva visión del tratamiento de la FPI con el abandono de los fármacos antiinflamatorios e inmunomoduladores y la utilización de fármacos antifibróticos17,18. La normativa de la Sociedad Española de Neumología y Cirugía Torácica (SEPAR) recomienda la pirfenidona como primera opción terapéutica actual en pacientes con enfermedad leve moderada2. No existen fármacos eficaces para la enfermedad grave.
N-acetilcisteínaLa N-acetilcisteína (NAC) es un potente antioxidante que modula la respuesta fibrótica. A pesar de ello, su indicación en FPI es controvertida. El estudio PANTHER descartó la indicación del tratamiento basado en la triple terapia (NAC+azatioprina+glucocorticoides), que se ha utilizado de forma habitual en el tratamiento de la FPI. Este estudio ha demostrado una mayor probabilidad de ingresos hospitalarios y una mayor mortalidad en los pacientes tratados con triple terapia frente a los tratados con NAC o placebo19. La eficacia de la NAC está siendo evaluada en la continuación del estudio PANTHER en el que se compara la NAC frente a placebo. No obstante, ante la ausencia de otras alternativas disponibles, puede utilizarse la NAC en monoterapia2.
Pirfenidona (Esbriet®)La pirfenidona posee propiedades antioxidantes, antifibróticas y antiinflamatorias. Diversos estudios han demostrado que su utilización reduce la progresión de la enfermedad en un 30%, disminuye de forma significativa el deterioro de la FVC y de la distancia recorrida en la prueba de la marcha de los 6 minutos, y aumenta el tiempo libre de síntomas. Es el único fármaco aprobado por la European Medicines Agency (EMA) para el tratamiento de la FPI leve-moderada, definida por FVC>50% y DLCO>35%. La pirfenidona es un fármaco bien tolerado. Los efectos secundarios más frecuentes son fotosensibilidad, molestias digestivas o alteraciones reversibles de la función hepática. Las contraindicaciones son hipersensibilidad al fármaco, uso de fluvoxamina, hepatopatía o nefropatía grave20,21.
NintedanibEs un inhibidor de la tirosinaquinasa que actúa sobre los factores de crecimiento endotelial vascular, plaquetario y fibroblástico. En un ensayo clínico en fase II, se ha observado que disminuye el deterioro de la FVC, disminuye el número de exacerbaciones de la enfermedad y mejora la calidad de vida. En la actualidad, están en desarrollo estudios en fase III con el fin de confirmar el efecto beneficioso del fármaco22.
Otros fármacosEn el momento actual se encuentran en fase de desarrollo ensayos clínicos con opciones terapéuticas farmacológicas vinculadas a la utilización de anticuerpos y antagonistas de factores de crecimiento. Otras opciones como las terapias celular y génica se encuentran en fase experimental.
Tratamiento no farmacológicoOxigenoterapia domiciliariaLa indicación de oxigenoterapia domiciliaria en pacientes con FPI e hipoxemia en reposo proviene, fundamentalmente, de extrapolar las conclusiones de los estudios realizados en pacientes con EPOC e insuficiencia respiratoria crónica. No hay datos concluyentes que avalen el empleo de oxigenoterapia de deambulación para pacientes que desaturan solo durante el esfuerzo, sin insuficiencia respiratoria en reposo. Ante la falta de datos concretos en pacientes con FPI, se recomienda administrar oxigenoterapia crónica domiciliaria ante la constatación de hipoxemia significativa en reposo o al final de la prueba de la marcha de los 6 minutos (SaO2 ≤88%)1,2.
Rehabilitación respiratoriaLa rehabilitación respiratoria mejora la respuesta al ejercicio y la calidad de vida aunque no se ha podido constatar efectos sobre la supervivencia a largo plazo.
Su beneficio es mayor y más sostenido cuando se realiza en pacientes en fase leve, por lo que la inclusión de los pacientes con FPI en programas de rehabilitación respiratoria debe hacerse en la fase más precoz posible23.
Trasplante pulmonarEs la única opción válida de tratamiento en pacientes en fases evolucionadas. Supone un aumento de la supervivencia y una mejora de la calidad de vida.
Los resultados obtenidos son inferiores a los referidos en pacientes trasplantados con EPOC, hipertensión pulmonar, fibrosis quística y otras enfermedades pulmonares24. Los criterios de inclusión para el trasplante se indican en la tabla 5.
Tratamiento de las complicaciones y comorbilidadesExacerbación agudaSe define como un rápido deterioro de la enfermedad en ausencia de infección respiratoria, insuficiencia cardíaca, embolismo pulmonar o cualquier otra causa identificable. Habitualmente cursa con agravamiento clínico (empeoramiento de la disnea en menos de 4 semanas) y afectación radiológica (opacidades de vidrio deslustrado o consolidaciones superpuestas a las imágenes de NIU en la TACAR). La evidencia para la recomendación de su tratamiento es muy escasa ante la ausencia de estudios concluyentes. El tratamiento más habitual es la utilización de bolos de corticoides a dosis altas (metilprednisolona 500-1.000mg/d) durante 3 días, continuado con dosis elevadas de prednisona (0,5mg/kg) asociados o no a azatioprina, ciclofosfamida o ciclosporina25.
Hipertensión pulmonarLa recomendación es la utilización de sildenafilo en pacientes que cursen con hipertensión pulmonar leve moderada constatada por cateterismo derecho26,27. No obstante, son precisos más estudios para evaluar la eficacia de los fármacos vasodilatadores.
Reflujo gastroesofágicoEs un factor de riesgo para la FPI por las frecuentes aspiraciones de contenido gástrico ácido. Se recomienda el tratamiento con medidas higiénico-dietéticas, inhibidores de la bomba de protones o cirugía antirreflujo si no fuera suficiente con las medidas anteriores28.
Tratamiento sintomático y cuidados paliativosLa tos debe tratarse para conseguir mejorar la calidad de vida y el descanso nocturno. Se recomienda el uso de codeína y glucocorticoides a dosis bajas. La disnea puede tratarse con dosis crecientes de morfina o sus derivados. Debe ofrecerse el tratamiento con cuidados paliativos para los pacientes en fase final de la enfermedad, tanto farmacológico para el control de síntomas como dolor, tos, disnea, depresión y ansiedad, como de apoyo psicoterápico, extensible este último para los familiares y cuidadores29. Es fundamental el soporte de asociaciones de pacientes. En España, existe la Asociación de Familiares y Enfermos de Fibrosis Pulmonar Idiopática (AFEFPI, www.afefpi.org).
FinanciaciónIntermune Spain. S.L.
Conflicto de interesesEn relación con el tema tratado en este documento:
Julio Ancochea declara haber recibido financiación por impartir conferencias en eventos educacionales y/o por asesoría científica y/o investigación Boehringer Ingelheim, InterMune, Actelion y Zambón.
Enrique Mascarós declara haber recibido financiación por asesoría para Intermune.
Jesús Molina declara haber recibido financiación por asesoría para Intermune.
José Antonio Quintano declara haber recibido financiación por asesoría para Intermune.
Juan Antonio Trigueros declara haber recibido financiación por asesoría para Intermune.
Antoni Xaubet declara haber recibido financiación por impartir conferencias en eventos educacionales y/o por asesoría científica y/o investigación de Intermune, Actelion, Almirall y GSK.
Consenso entre las Sociedades Españolas de Atención Primaria con la asesoría del Programa Integrado de Investigación de Enfermedades Pulmonares Intersticiales Difusas de la Sociedad Española de Neumología y Cirugía Torácica (SEPAR).
