




La miastenia gravis es una enfermedad autoinmune en la que se sintetizan autoanticuerpos contra el receptor nicotínico para la acetilcolina en la unión neuromuscular. Su incidencia es de 3–4 casos por millón de habitantes y año, por lo que es rara su presentación en las consultas de atención primaria. El síntoma principal es la debilidad muscular fluctuante. La mitad de los pacientes presentan síntomas oculares como la diplopía o la ptosis palpebral. Presentamos el caso clínico de una mujer de 19 años que presentó estos síntomas de forma lenta y progresiva. La aparición insidiosa de los síntomas dificultó el diagnóstico inicial de esta enfermedad. Sin embargo, el carácter longitudinal de la asistencia en atención primaria nos permitió hacer una correcta historia clínica y detectar los cambios en la exploración neurológica que nos llevaron a derivar a la paciente a un servicio de neurología de forma urgente.
Myasthenia gravis (MG) is an autoimmune disease of the neuromuscular junction in which there are antibodies against the nicotinic acetylcholine receptor. MG incidence is about 3–4 cases per one million inhabitants/ year, so that it is rarely seen in primary care. Fluctuating muscle weakness is the main symptom of this disease. Half of population show ocular-associated symptoms as diplopia and palpebral ptosis. We are presenting the clinical case of a 19-year old woman who gradually and progressively developed these symptoms. The insidious development of the disease made its diagnosis difficult. However, the long-term attendance in primary care made it possible for the physicians to obtain a correct clinical history and to detect the changes through neurological examination that lead to the urgent referral of the patient to the Neurology Department.
La miastenia gravis (MG) es una enfermedad autoinmune en la que se sintetizan autoanticuerpos contra el receptor nicotínico para la acetilcolina a nivel de la unión neuromuscular. Su incidencia es de 3–4 casos por millón de habitantes y año, por lo que es rara su presentación en las consultas de atención primaria. Se presenta dos veces más en mujeres que en hombres y la edad de inicio muestra dos picos: entre la 2.a y la 4.a décadas de la vida y entre la 6.a y la 8.a. En un 5% de los pacientes se asocian otras enfermedades autoinmunes, como la artritis reumatoide, el lupus eritematoso sistémico o la anemia perniciosa y en un 10% la enfermedad tiroidea autoinmune. El 10–15% de los enfermos presenta un timoma y el 50–70% de los casos una hiperplasia tímica, por lo que es obligatoria la realización de una TAC torácica.
El síntoma principal de la MG es la debilidad muscular, que empeora con el ejercicio y mejora con el reposo. La mitad de los pacientes presenta síntomas oculares como la diplopía o la ptosis palpebral. El hecho de que estos síntomas se presenten de forma fluctuante y empeoren con la fatiga puede dificultar la sospecha clínica en las etapas iniciales y demorar el diagnóstico. El carácter longitudinal de la asistencia en atención primaria nos permitirá hacer una correcta historia clínica y favorecerá la detección de los cambios en la exploración neurológica característicos de la evolución de esta enfermedad.
Presentamos el caso de una mujer de 19 años de edad, sin antecedentes personales ni familiares de interés, que consulta inicialmente por ptosis palpebral y debilidad muscular fluctuante en miembros inferiores de varias semanas de evolución. Queremos destacar que una buena anamnesis y exploración física permiten plantear la hipótesis diagnóstica de una enfermedad poco frecuente, que requiere un tratamiento específico y un seguimiento a largo plazo.
Caso clínicoSe trata de una mujer de 19 años de edad, soltera, que vive con su madre, es estudiante y trabaja como camarera los fines de semana en una discoteca. No presentaba antecedentes médicos ni quirúrgicos de interés ni tomaba ningún tratamiento. Acudió por primera vez a nuestra consulta en agosto de 2008 por astenia y ptosis palpebral, que describía como dificultad para la elevación palpebral bilateral, de varias semanas de evolución y que no interfería en su actividad diaria.
En la exploración física, se apreció buen estado general, con buena coloración de piel y mucosas. Tensión arterial: 95/65mmHg; Frecuencia cardíaca: 70lpm. No se apreciaban bocio ni adenopatías en la exploración cervical y las carótidas eran rítmicas y simétricas, sin soplos. La auscultación cardiorrespiratoria era normal y el abdomen blando y depresible y las extremidades inferiores normales. En la exploración neurológica únicamente se apreciaba una leve ptosis palpebral bilateral tras varios parpadeos, con visión conservada y pupilas isocóricas y simétricas. La fuerza, la sensibilidad y los reflejos osteotendinosos en miembros superiores e inferiores estaban conservados. Los signos meníngeos eran negativos y la marcha era normal.
Exploraciones complementarias solicitadas en atención primaria y evoluciónSe solicitó analítica de sangre. En la bioquímica general, se apreció hipoglucemia de 57 ng/dl con ácido úrico, calcio, colesterol y triglicéridos, creatinina, hierro sérico, glucosa,transaminasa glutámico oxalacética(GOT, del inglés glutamic oxaloacetic transaminase) y transaminasa glutámico pirúvica (GPT, del inglés glutamic pyruvic transaminase), proteínas totales y ferritina normales. La hematimetría fue normal. La hormona tiroestimulante (TSH, del inglés thyroid-stimulating hormone) se encontraba dentro de límites normales.
En octubre de 2008 la paciente acudió de nuevo a consulta refiriendo episodios de claudicación mandibular tras unos minutos de masticación, sin alteraciones en la deglución, y disminución de fuerza en miembros inferiores bilateral que se manifestaba al subir un tramo de escaleras. Asimismo, refirió persistencia de dificultad para la elevación palpebral bilateral y aparición de visión doble de forma episódica. En la exploración neurológica se encontraba alerta, consciente y orientada. Las pupilas eran normales. Presentaba aumento de ptosis palpebral con respecto a la exploración previa con diplopía en la dextroversión, que mejoraba con la visión monocular. La fuerza era simétrica y conservada. Sin alteraciones de la sensibilidad. Presentaba fatiga en miembros inferiores con siete sentadillas y conservaba fuerza en miembros superiores. La marcha seguía siendo normal.
Solicitamos interconsulta preferente con especialista en Neurología y recomendamos reposo. La paciente acudió cinco días después a nuestra consulta por presentar disminución de fuerza en miembros superiores, precisando ayuda de su madre para vestirse. Realizamos una nueva exploración neurológica en la que se apreciaba fatiga de miembros superiores tras elevarlos diez veces, además de los hallazgos exploratorios previos. En ese momento derivamos a la paciente a urgencias hospitalarias para valoración por el neurólogo de guardia.
La paciente ingresó en el Servicio de Neurología para estudio. Le realizaron analítica de sangre con bioquímica normal, creatinfosfocinasa (CPK, del inglés creatine phosfhokinase) 47 U/l, proteinograma normal, TSH de 1,06 mU/l, hemograma normal, autoanticuerpos no organoespecíficos (ANOE; incluye anticuerpos antinucleares [ANA]; anticuerpos antimúsculo [ASMA] liso; anticuerpos antimitocondriales [AMA]) negativos, anticuerpos antitiroideos negativos, factor reumatoide normal, cuantificación de inmunoglobulinas normal, anticuerpos antirreceptor de acetilcolina, fenotipo locus del antígeno de histocompatibilidad (HLA, del inglés histocompatibility locus antigen): normales.
Ante la sospecha de MG se realizó el test de la piridostigmina, administrando a la paciente un comprimido de bromuro de piridostigmina de 60mg, con mejoría significativa de la ptosis palpebral y claudicación mandibular.
Para confirmar el diagnóstico, se realizó un electromiograma de fibra aislada. Esta prueba electromiográfica consiste en el empleo de un electrodo de aguja con una superficie activa muy pequeña, para recoger el potencial de dos fibras musculares cercanas pertenecientes a la misma unidad motora. Normalmente, existe una pequeña variación en el intervalo de tiempo entre la aparición de uno y otro potencial, lo que se denomina jitter. En los pacientes con miastenia se produce un claro aumento del jitter con densidad de fibras normal. Este resultado fue objetivado en el caso de nuestra paciente, indicando una alteración de la transmisión neuromuscular a nivel de la placa motora, compatible con MG.
Tras el diagnóstico se realizó una TAC torácica en la que se apreciaron restos tímicos de volumen normal para el grupo de edad de la paciente, sin objetivarse imagen característica de timoma.
TratamientoEl tratamiento de los pacientes con MG deber ser individualizado y deben considerarse en primer lugar los inhibidores de la acetilcolinesterasa. El fármaco más frecuentemente utilizado como tratamiento de mantenimiento a largo plazo es el bromuro de piridostigmina. La dosis de inicio recomendada es de 30mg/6–8h, que se aumenta según la respuesta del paciente hasta una dosis máxima de 120mg/4h.
Los esteroides (prednisona) obtienen una respuesta clínicamente favorable en al menos el 70% de los pacientes. La dosis inicial recomendada es de 1mg/kg/día, pero se suele comenzar con dosis menores (en torno a 20–30mg/día) e ir ascendiendo progresivamente porque durante los primeros 10–15 días de tratamiento esteroideo puede producirse un empeoramiento clínico del paciente. La dosis alcanzada debe mantenerse durante varios meses, para posteriormente intentar una suspensión muy gradual, o dejar al paciente con la dosis mínima eficaz.
La siguiente línea de tratamiento son los inmunosupresores (azatioprina y ciclosporina). La azatioprina es el más frecuentemente utilizado. La dosis es de 2–3mg/kg/día, comenzando con 50mg/día y aumentando progresivamente. Sus efectos no son apreciables hasta 4–6 meses. Además, hay que tener en cuenta que es potencialmente teratogénica y que puede producir hepatotoxicidad y mielotoxicidad, por lo que son necesarios controles analíticos estrictos, sobre todo al inicio del tratamiento.
En el caso de nuestra paciente, se inició tratamiento durante el ingreso con prednisona 75mg al día, bromuro de piridostigmina 30mg cada 8h y omeprazol 20mg al día. La paciente presentó buena tolerancia al tratamiento y fue dada de alta hospitalaria; se la derivó a cirugía donde meses más tarde realizaron extirpación del timo. El seguimiento del tratamiento de mantenimiento fue realizado desde nuestra consulta de atención primaria, realizando tratamiento con una dosis descendente de prednisona y la misma dosis de bromuro de piridostigmina hasta nueva valoración por especialista en neurología de forma ambulatoria.
Discusión del casoHasta llegar a la sospecha clínica de MG, la paciente presentó una evolución lenta de los síntomas en los primeros dos meses que fue más rápida en el último mes, pudiendo clasificarse dentro de una miastenia generalizada.
En el diagnóstico diferencial de la MG se deben considerar otros trastornos que causan debilidad de la musculatura craneal y somática; entre ellos cabe citar el síndrome miasténico de Lambert-Eaton (SMLE), la neurastenia, el hipertiroidismo, el botulismo, la presencia de masas intracraneales y la oftalmoplejía externa progresiva.
El SMLE es un trastorno presináptico de la unión neuromuscular en la que se producen autoanticuerpos contra los canales del calcio tipo P/Q. Esto puede causar una debilidad similar a la que se produce en la MG. La musculatura proximal de los miembros inferiores está afectada con frecuencia, aunque también pueden estar afectados otros grupos musculares. Hasta en un 70% de los pacientes se observan alteraciones de pares craneales, como ptosis palpebral y diplopía, características también observadas en la MG. No obstante, ambas afecciones pueden diferenciarse fácilmente, ya que los pacientes con SMLE muestran disminución o ausencia de los reflejos, alteraciones del sistema nervioso autónomo, como sequedad de boca e impotencia, así como respuestas de incremento tras la estimulación nerviosa repetitiva.
La neurastenia puede causar debilidad y fatiga, aunque en el estudio del músculo habitualmente se observa la “liberación espasmódica” característica de los trastornos no orgánicos, y la fatiga en estos pacientes implica cansancio o apatía en lugar de una disminución de la fuerza muscular tras el esfuerzo repetido.
El hipertiroidismo se diagnostica o excluye fácilmente mediante las pruebas de función tiroidea, que se deben realizar sistemáticamente en los pacientes con sospecha de MG.
El botulismo puede producir esta clase de debilidad, aunque en estos casos las pupilas suelen estar afectadas y la estimulación nerviosa repetitiva determina una respuesta de incremento y no de disminución.
Las masas intracraneales a veces pueden causar una diplopía que simula los síntomas de la MG, debido a la compresión de los nervios que inervan la musculatura extraocular (p. ej. meningioma de la cresta esfenoidal), aunque habitualmente una resonancia magnética del cráneo y de las órbitas demuestra la lesión.
La oftalmoplejía progresiva externa es un trastorno que produce debilidad de la musculatura extraocular, y que se puede acompañar de debilidad en la musculatura proximal de los miembros y de otras alteraciones sistémicas. La mayoría de los pacientes con este trastorno muestran alteraciones mitocondriales que pueden ser detectadas en la biopsia muscular, tras descartar MG mediante las pruebas correspondientes citadas con anterioridad.
Actualmente, el pronóstico de la enfermedad con los tratamientos existentes es bueno. La mortalidad por crisis miasténicas es menor del 5%, gracias a los avances en cuidados intensivos y a la inmunoterapia.
El curso de la enfermedad es variable, sobre todo dentro del primer año de presentación. Casi el 85% de los pacientes con síntomas oculares iniciales pueden desarrollar debilidad muscular fluctuante dentro de los tres primeros años. Las crisis miasténicas suceden en aproximadamente el 20% de los pacientes en el primer año de la enfermedad y se definen como la debilidad progresiva de fuerza que desemboca en insuficiencia ventilatoria por fallo muscular del diafragma o por debilidad orofaríngea. Constituyen una urgencia médica, que requiere ventilación mecánica en una unidad de cuidados intensivos.
Seguimiento de la enfermedadAunque el seguimiento de la enfermedad es compartido con el especialista en neurología de forma periódica, el médico de familia es quien establecerá una relación más cercana con el paciente y le verá con mayor asiduidad, incluso por motivos de consulta distintos de la MG.
Por ello, se debe tener en cuenta que los síntomas de la MG pueden empeorar con algunas enfermedades como infecciones de vías respiratorias altas, embarazo, hipotiroidismo e hipertiroidismo y síntomas como la fiebre.
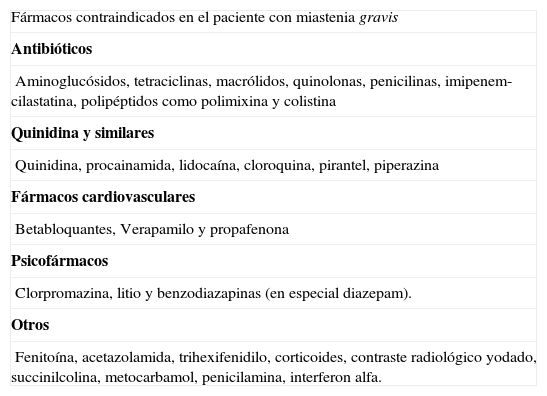
Además, existen bastantes fármacos que están contraindicados en estos pacientes y que siempre debe tener en cuenta su médico de atención primaria antes de iniciar el tratamiento de otras patologías (tabla 1).
Fármacos que pueden empeorar una miastenia gravis e incluso provocar una crisis miasténica
| Fármacos contraindicados en el paciente con miastenia gravis |
| Antibióticos |
| Aminoglucósidos, tetraciclinas, macrólidos, quinolonas, penicilinas, imipenem-cilastatina, polipéptidos como polimixina y colistina |
| Quinidina y similares |
| Quinidina, procainamida, lidocaína, cloroquina, pirantel, piperazina |
| Fármacos cardiovasculares |
| Betabloquantes, Verapamilo y propafenona |
| Psicofármacos |
| Clorpromazina, litio y benzodiazapinas (en especial diazepam). |
| Otros |
| Fenitoína, acetazolamida, trihexifenidilo, corticoides, contraste radiológico yodado, succinilcolina, metocarbamol, penicilamina, interferon alfa. |
Este es un caso en el que la longitudinalidad que ofrece la consulta de atención primaria cobra gran importancia, pues nos permitió ir encadenando los síntomas, los signos de la exploración neurológica, los resultados de las pruebas, la derivación al especialista adecuado, etc. creando una historia clínica que fue fundamental en el diagnóstico final de MG.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

Medicina de Familia. SEMERGEN sigue las recomendaciones para la preparación, presentación y publicación de trabajos académicos en revistas biomédicas