






El síndrome de Steinert o distrofia miotónica tipo I es la distrofia muscular más frecuente del adulto. Se trata de una enfermedad muscular autosómica dominante que presenta el fenómeno de anticipación. La anomalía molecular está causada por una expansión de repeticiones del trinucleótido que contiene citosina-timidina-guanosina en el gen DMPK (dystrophia miotónica proteincinasa) localizado en la región cromosómica 19q13.3. Presentamos el caso de un varón de 39 años con rasgos físicos característicos del síndrome de Steinert. Revisamos su historia natural y encontramos un origen congénito del mismo, llegamos hasta su bisabuela buscando el origen hereditario de su enfermedad. Dada la evolución del paciente, y debido a que se trata de una enfermedad con afectación multiorgánica, consideramos que los médicos de Atención Primaria deben desempeñar un papel clave en el control y seguimiento de los pacientes con síndrome de Steinert.
Steinerts syndrome or myotonic dystrophy type I is the most frequent muscular dystrophy in the adult. It is an autosomal dominant muscular disease that exhibits the anticipation phenomenon. The molecular abnormality is caused by an expansion of a cytosine-thymine-guanine (CTG) trinucleotide repeats of the DMPK gene (Myotonic dystrophy protein kinase) located on chromosome 19q13.3. We present the case of a 39-year old man with typical clinical features of Steinerts disease. After reviewing his natural history, we found a congenital origin and of it, the hereditary origin of the disease going back to his great-grandmother. Because of the evolution of our patient and because Steinerts disease is a multi-organ disease, we consider that the family doctors should play a key role in the control and follow-up of patients with Steinert´s disease.
Presentamos a un varón de 39 años que presenta una calvicie frontal, cara alargada con pómulos hundidos y ptosis palpebral (fig. 1). Observamos atrofia y debilidad muscular en la cara, el cuello, el antebrazo, las manos y los pies. El desarrollo de los caracteres sexuales primarios y secundarios es adecuado. Presenta un retraso mental leve, siendo capaz de leer, escribir, sumar y restar en el momento actual. Refiere hipersomnia y somnolencia diurna, así como episodios diarreicos frecuentes. Es prácticamente dependiente para las actividades de la vida diaria. En el momento actual vive con su tía y trabaja de conserje en una empresa familiar.

Figura 1. Nuestro paciente a la derecha y su madre a la izquierda.
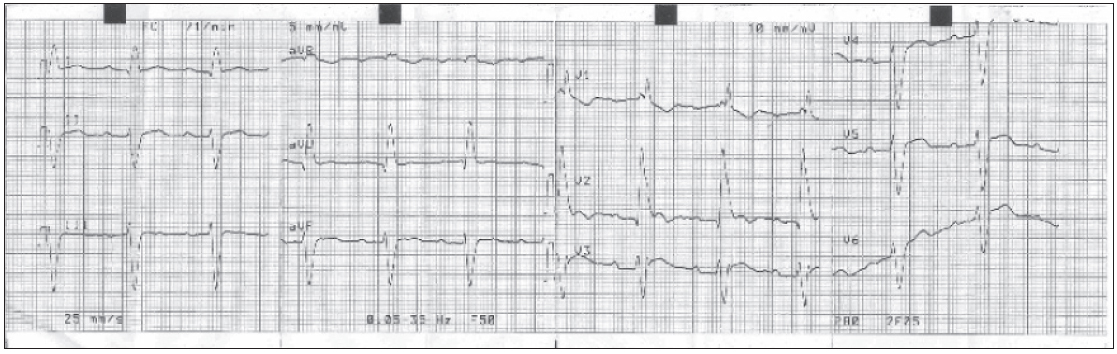
En la analítica de sangre destacamos un colesterol total de 280 mg/dl, colesterol LDL de 189 mg/dl, creatincinasa de 236 UI/l y testosterona de 2,76 ng/ml, siendo el resto normal. El electrocardiograma (ECG) muestra un bloqueo de la rama derecha, un hemibloqueo anterior izquierdo y un bloqueo aurículo ventricular de primer grado (fig. 2).
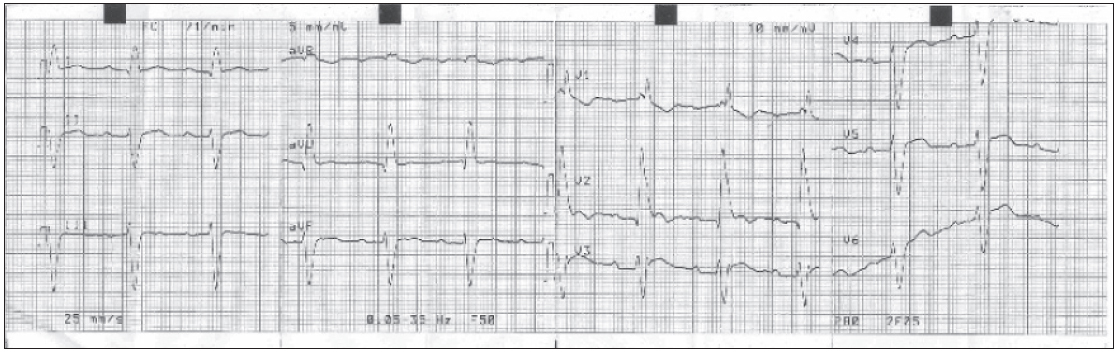
Figura 2. Electrocardiograma.
Como antecedentes personales encontramos que al nacer pesó 3.300 g y tuvo un periodo neonatal crítico, por lo que necesitó ser ingresado en la Unidad de Cuidados Intensivos por distrés respiratorio, hipotonía, dificultad para la deglución y succión débil. Tuvo que ser intervenido de un absceso cutáneo craneal. Tras un mes de estancia fue dado de alta con un peso de 1.500 g. Inició la marcha a los 16 meses y fue intervenido poco después de pies zambos y a los 6 años necesitó una reintervención quirúrgica. Fue intervenido por criptorquidia bilateral y hernia inguinal derecha a los 6 años. Presentó cataratas desde los 16 años y a los 25 se le implantaron lentes intraoculares. A los 26 años sufrió un síncope, por lo que fue ingresado en la unidad coronaria sin causa filiada, y a los 28 se observó una taquicardia ventricular monomorfa sostenida por reentrada rama-rama que requirió ablación por radiofrecuencia e implantación de un marcapasos.
Al investigar sobre sus antecedentes familiares encontramos que su madre presentó una facies miotónica (fig. 1), hipersomnia y ronquidos, caídas frecuentes, debilidad muscular y colon irritable. Fue intervenida de cataratas a los 30 años y presentó varias neumonías en su vida, la primera a los 18 años y la última a los 60 años, a consecuencia de la cual falleció. Su madre tenía 2 hermanas, una falleció a los 3 meses de muerte súbita y la otra, libre de patología, es la que convive con nuestro paciente. El abuelo materno fue el segundo de cinco hermanos. Fue diagnosticado de cataratas a los 48 años y falleció a los 65 años por una arritmia no filiada. Sus hijas referían que sufría presíncopes habituales y que era un hombre con debilidad muscular, con 30 años era incapaz de abrir una lata. Nos entregaron una foto, en la que podemos observar un gran parecido físico con nuestro paciente (fig. 3). Finalmente la bisabuela materna, madre de su abuelo, fue descrita como una persona con debilidad muscular y que falleció a los 40 años por una neumonía.
Se realizó un electromiograma que era compatible con una distrofia miotónica de Steinert. En el año 2006 se realizó el estudio genético de nuestro paciente y de su madre, que mostró una expansión de ADN de aproximadamente 1.000 repeticiones de citosina-timidina-guanosina (CTG) en el extremo 3´UTR del gen DMPK (dystrophia miotónica proteincinasa), por lo que son portadores de la enfermedad de Steinert.

Figura 3. El abuelo.
DISCUSIÓNEl síndrome de Steinert, o distrofia miotónica tipo I, es una enfermedad muscular autosómica dominante. La anomalía molecular está causada por una expansión de repeticiones del trinucleótido que contiene CTG en el gen DMPK localizado en la región cromosómica 19q13.3. Clínicamente se caracteriza por miotonía (dificultad para relajar un músculo contraído) y una afectación multiorgánica que incluye diferentes grados de debilidad muscular, trastornos de conducción cardiaca, trastornos endocrinos, respiratorios, oculares y neurológicos, así como una calvicie precoz.
La prevalencia de la distrofia miotónica es de uno por cada 8.000 individuos en la población general, siendo el síndrome de Steinert o distrofia miotónica tipo I la distrofia muscular más frecuente del adulto1. Esta enfermedad genética presenta el fenómeno de anticipación, que se define como el inicio progresivamente más precoz y con mayor intensidad de los síntomas en las generaciones siguientes.
La edad de inicio de la enfermedad varía desde el periodo neonatal hasta la sexta o séptima década de la vida, y es entre los 20 y 40 años cuando la aparición de la enfermedad es más frecuente. El pronóstico varía con la edad de aparición, y es más grave cuanto antes comience la enfermedad. En el periodo neonatal fallecen hasta el 16% de los casos1,2. De igual forma, cuantas más copias de CTG haya, antes se manifestará la enfermedad y más grave será la misma3. En general, por encima de 750 copias existe más riesgo de manifestación congénita4.
La transmisión de la distrofia miotónica de Steinert se realiza de manera autosómica dominante, si bien es cierto que la forma congénita se transmite casi exclusivamente por vía materna5. Debido a esto, el caso de nuestro paciente y el de su abuelo, que probablemente transmitió la enfermedad, son aún más interesantes.
La miotonía y la debilidad muscular son las manifestaciones que más definen a la enfermedad. La miotonía consiste en la dificultad para relajar un músculo contraído, clásicamente era observada por el médico al dar la mano al paciente, el cual presentaba un retardo en la relajación de la misma al concluir el saludo. La debilidad de los músculos faciales y temporales constituyen la “facies miopática”, siendo muy característica la ptosis palpebral, como podemos observar en las fotografías. Los otros grupos musculares afectados son los cervicales y la musculatura de los miembros, con predominio distal6, pudiendo ocasionar un pie equino como en nuestro paciente, y una impotencia funcional de los músculos intrínsecos de la mano como en su abuelo. En los ojos la manifestación más característica son las cataratas7. En este caso podemos observar el fenómeno de anticipación, ya que la aparición de las mismas en el abuelo, en la madre y en nuestro paciente se produce a los 48, 30 y 16 años respectivamente.
La insuficiencia gonadal es frecuente en los hombres, y es responsable de una oligospermia o azoospermia8. En ocasiones encontramos la presencia de criptorquidia. En las mujeres provoca amenorrea, dismenorrea y quistes ováricos. También son frecuentes los trastornos del metabolismo hidrocarbonato por resistencia a la insulina y trastornos lipídicos. El patrón de calvicie frontal suele ser un rasgo característico.
Son características las alteraciones de conducción con arritmias9, así como los trastornos de conducción auriculoventriculares e intraventriculares que aparecen de forma progresiva10. Si bien es cierto que las arritmias más frecuentes son la fibrilación y el flúter auricular, las más graves son las ventriculares, pudiendo aparecer taquicardias ventriculares con un mecanismo de reentrada10, como sucede en el caso de nuestro paciente y posiblemente en el de su abuelo. El riesgo de muerte súbita es elevado, se estima que está entre el 4 y el 10%2. El ECG y el holter son pruebas complementarias obligadas para la detección de dichas alteraciones10-12.
Los trastornos digestivos más frecuentes son disfagia, pirosis, regurgitación dispepsia, trastornos en el ritmo abdominal y dolor abdominal. En ocasiones pueden ser la manifestación inicial de la distrofia13. En el 50% de los pacientes se puede observar una elevación de la gammaglutamil transpeptidasa14.
La hipersomnia y la somnolencia diurna con o sin apnea del sueño son una característica frecuente15, por lo que se recomienda hacer estudios polisomnográficos16. Los trastornos respiratorios son habituales, por lo que suelen presentar debilidad diafragmática y aspiraciones bronquiales por insuficiencia de la musculatura orofaríngea en la deglución.
En general, la aparición de déficit cognitivo es más importante cuanto antes aparezca la enfermedad. El retraso mental en las formas congénitas es una característica habitual17.
La muerte se produce en la quinta o sexta década de la vida, y las causas más frecuentes son la debilidad muscular progresiva y las arritmias cardiacas. Los pacientes fallecen por neumonía, insuficiencia cardiaca o enfermedad intercurrente1,18.
En nuestro paciente y en sus familiares coinciden muchas de las características clínicas anteriormente descritas. Así pues, podemos explicar los trastornos padecidos por los ancestros de nuestro paciente remontándonos hasta su bisabuela. Si nos centramos en nuestro paciente, su historia comenzó en el periodo neonatal con la presencia de distrés respiratorio, debilidad e hipotonía, por lo que presuponemos un origen congénito de la enfermedad. A medida que su vida transcurre podemos observar la aparición de rasgos de la miopatía Steinert tan leves como las cataratas y tan graves como las arritmias ventriculares. Pero no es hasta el año 2006 cuando obtuvimos la confirmación diagnóstica mediante el estudio genético, esto nos animó a realizar una investigación de la historia familiar del paciente, encontrando una probable línea genética que nos lleva hasta su bisabuela.
La miopatía de Steinert es una enfermedad multiorgánica que va a requerir una visión global del paciente, y es en las consultas de Atención Primaria donde muchos de los problemas serán diagnosticados y resueltos, siempre en colaboración con un neurólogo.
Correspondencia: A.L. Aguilar Shea.
Centro de Salud Manzanares. Consultorio El Bolao
C/Peña Hoyuela 16.
28413 El Bolao. Madrid. España.
Correo electrónico: antonioaguilarshea@gmail.com
Recibido el 20-05-08; aceptado para su publicación el 17-09-08.
