



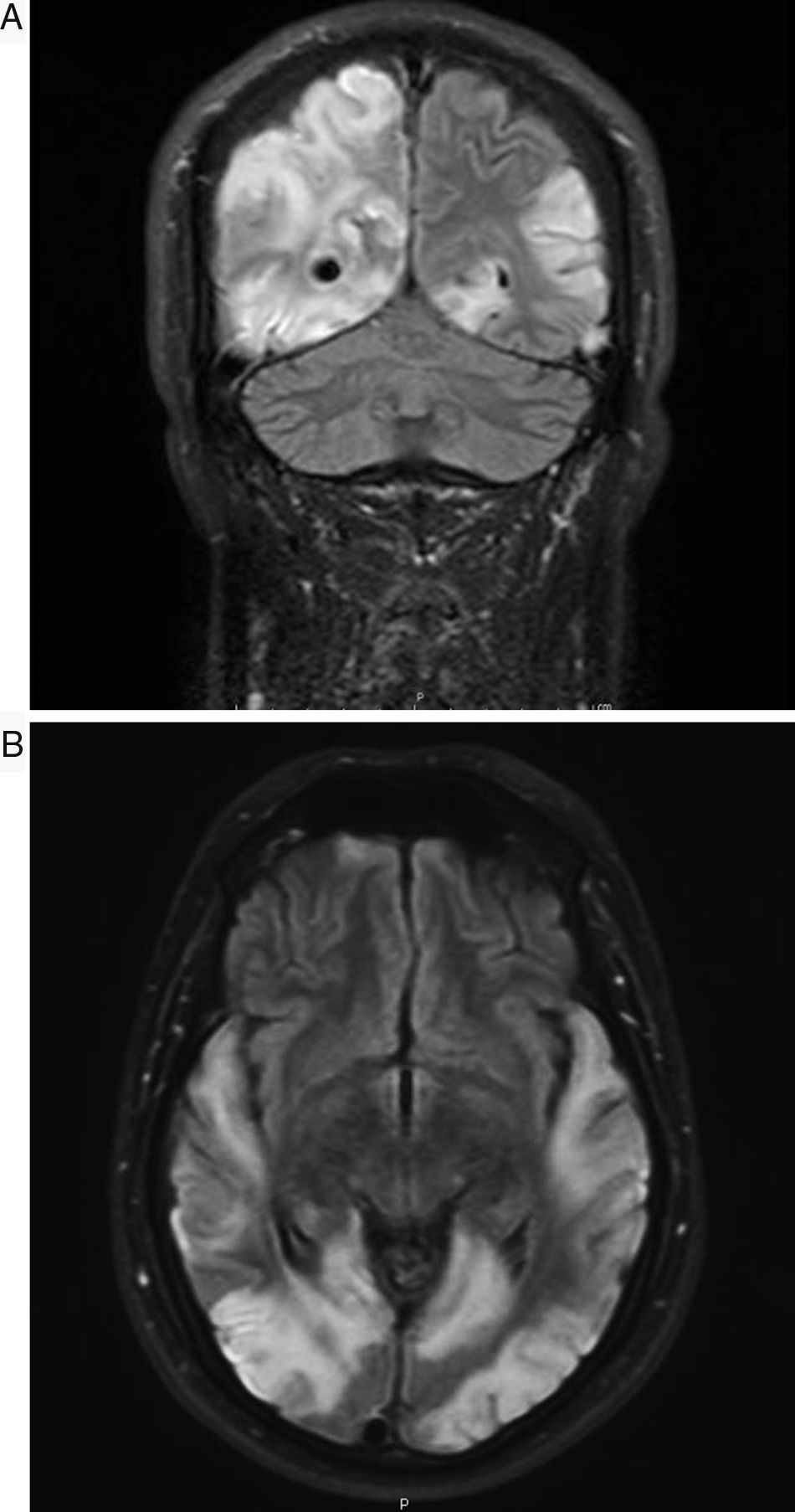
El síndrome de MELAS forma parte de las denominadas enfermedades mitocondriales, siendo un problema fundamental el no existir series largas de pacientes con el mismo defecto molecular y la misma manifestación clínica que permitan realizar estudios concluyentes sobre la efectividad de los diversos fármacos aplicados1. Presentamos el caso de un varón de 30 años, sin antecedentes de interés, que en enero de 2013 presentó episodio compatible con crisis epiléptica focal secundariamente generalizada, con posterior status epiléptico que precisó ingreso en la UVI. Se realizó, entonces, resonancia magnética (RM) craneal dónde se apreció únicamente engrosamiento cortical focal de giro lingual izquierdo sugiriendo como primera posibilidad diagnóstica displasia cortical focal. El paciente estuvo asintomático hasta principios de septiembre de 2014, cuando fue ingresado en nuestro centro tras haber presentado un cuadro brusco de pérdida de visión en hemicampo izquierdo. En una primera valoración neurológica presentaba en la exploración únicamente hemianopsia homónima izquierda y discreta dismetría en maniobra dedo-nariz en miembro superior izquierdo. El paciente sufrió un llamativo y rápido empeoramiento durante los días siguientes, desarrollando ceguera cortical, sordera cortical y afasia completa. Presentaba importante inquietud psicomotriz, en ocasiones agitación franca, con jergafasia continua y actos inmotivados. Su única forma de contacto con el exterior era mediante el tacto y reconocía a su mujer por el anillo que ella llevaba. Analíticamente destacaba una elevación de lactato tanto en sangre (2,7mM/l) como en líquido cefalorraquídeo (3,36mM/l), así como elevación de L-carnitina y carnitina total en sangre. La creatincinasa fue normal. No presentaba datos de miopatía en el electromiograma y la audiometría mostró la presencia de una hipoacusia neurosensorial bilateral leve. En una nueva RM se objetivó extensión importante de las lesiones con afectación cortical hemisférica izquierda (predominio occipital, parietal y temporal), hiperintensidades en secuencias T2 y FLAIR, con edema y zonas de efecto masa, así como restricción en la difusión. Presentaba también afectación hemisférica derecha occipito-parietotemporal de similares características, y alguna zona de necrosis cortical laminar (figs. 1 y 2A y B). La biopsia muscular mostró cambios inespecíficos y ausencia de cambios morfológicos diagnósticos de miopatía mitocondrial (no fibras rojo rasgadas, no fibras COX-negativas). El estudio genético fue positivo para la mutación A3243G. Durante su ingreso recibió tratamiento con fenitoína 100mg en pauta 100-50-100, levetiracetam 1.000mg/12h, clonazepam 0,5mg/12h, ubiquinol 200mg/8h, idebenona 90mg/8h, arginina 6g/8h, complejos vitamínicos (B2 riboflavina 50mg/8h, B1 Tiamina® 300mg/medio comprimido/12h, vitamina E 200mg/un comprimido al día/lunes, miércoles y viernes; vitamina C 2g al día). Se objetivó cierta mejoría evolutiva. Este caso presenta varios aspectos de especial interés. En primer lugar, la edad de presentación tardía, lo cual sucede en alrededor de un 20% de los casos. Destaca también el hecho de que el paciente no presentara afectación muscular. La afectación muscular es especialmente frecuente en pacientes portadores de la mutación A3243G. Distintos estudios han revelado que la mutación A3243G produce un defecto severo combinado de la cadena respiratoria en los mioblastos. En la biopsia muscular suelen encontrarse depósitos grasos y es habitual la presencia de fibras rojo rasgadas. El porcentaje de pacientes diagnosticados de MELAS (independientemente de la mutación) con biopsia negativa ronda el 10% según algunos trabajos2. También resulta de interés la evolución tan agresiva que presentó el cuadro, pasando en apenas 3 días de presentar únicamente una hemianopsia a un estado de ceguera, sordera y afasia mixta que imposibilitó de manera prácticamente total la comunicación del paciente con el medio. La sordera de origen central ha sido descrita muy raramente3. En lo referente al tratamiento recibido, los complejos vitamínicos (B2, B1, E y C) en este caso actuarían como sustancias antioxidantes, lo que parece útil para corregir el daño oxidativo, aunque tampoco existen estudios concluyentes sobre su efectividad4. Como dato a destacar además se inició tratamiento con L-arginina, precursor del óxido nítrico cuyos niveles han sido analizados en distintos trabajos, encontrándose una disminución de los mismos tanto en periodo agudo como en fase interictal en comparación con sujetos control. La arginina se considera uno de los fármacos más prometedores5,6 y, aunque sus mecanismos de actuación no son completamente conocidos, parece que tiene efecto en la regulación vascular, produciendo vasodilatación mediante aumento del óxido nítrico. Esto produciría un aumento de la capacidad aeróbica y una mejoría en el metabolismo muscular6. La administración se puede realizar vía infusión intravenosa en fase aguda, con una actuación rápida en menos de 24h, con mejoría sintomática. Sin embargo, los síntomas pueden empeorar en los días sucesivos si no se continúa una suplementación vía oral, con una dosis recomendada de unos 0,5g/kg/día. En la actualidad nuestro paciente recibe una dosis vía oral de arginina de 6g/8h. El paciente permaneció ingresado durante aproximadamente mes y medio, objetivando cierta mejoría evolutiva. Al alta obedecía una orden sencilla y era capaz de elaborar frases cortas y coherentes. Presentó discreta mejoría de la visión, actualmente distingue colores, formas y movimiento. Persiste déficit de lenguaje con parafasias, por lo que continúa realizando logopedia ambulatoria. En ningún momento se ha objetivado déficit motor. Mantiene tratamiento con complejos vitamínicos, arginina vía oral 6g/8h, clonazepam 0,5mg/8h, levetiracetam 500mg/12h y lacosamida 100mg/8h, habiéndose suspendido la fenitoína. Se mantiene estable y no ha vuelto a precisar nuevos ingresos hasta el momento.
Compartir
 Coronal). B) Axial. Lesiones hiperintensas en secuencia de resonancia FLAIR afectando al cortex temporo-parieto-occipital bilateral, que no se corresponden con territorios vasculares.")

