



En los últimos años se ha observado que moléculas como el péptido relacionado con el gen de la calcitonina (CGRP) y, en menor grado, el péptido activador de la adenilato-ciclasa pituitaria estaban elevadas durante los ataques de migraña y en la migraña crónica tanto en líquido cefalorraquídeo como en suero y que su reducción farmacológica tenía una significación clínica con una mejoría en la migraña de los pacientes. Es lógico por tanto que una de las principales líneas de investigación en migraña se base en el papel del CGRP en la fisiopatología de esta entidad.
DesarrolloDesde el Grupo de Estudio de Cefaleas de la Sociedad Española de Neurología nos planteamos la redacción de este documento, cuyo objetivo es abordar, basándonos en la evidencia publicada, cuestiones tan importantes como el papel del CGRP en la fisiopatología de la migraña, el mecanismo de acción de los anticuerpos monoclonales y de los gepantes, el análisis crítico de los resultados de los diferentes estudios, el perfil del paciente que podría ser candidato al tratamiento con anticuerpos monoclonales y su impacto en términos de farmacoeconomía.
ConclusionesEl desarrollo clínico de los gepantes, antagonistas del CGRP, para el tratamiento agudo del ataque de migraña y de los anticuerpos monoclonales contra ligando y contra el receptor del CGRP ofrecen resultados esperanzadores para nuestros pacientes.
It has been observed in recent years that levels of such molecules as calcitonin gene–related peptide (CGRP) and, to a lesser extent, the pituitary adenylate cyclase–activating peptide are elevated during migraine attacks and in chronic migraine, both in the cerebrospinal fluid and in the serum. Pharmacological reduction of these proteins is clinically significant, with an improvement in patients’ migraines. It therefore seems logical that one of the main lines of migraine research should be based on the role of CGRP in the pathophysiology of this entity.
DevelopmentThe Spanish Society of Neurology's Headache Study Group decided to draft this document in order to address the evidence on such important issues as the role of CGRP in the pathophysiology of migraine and the mechanism of action of monoclonal antibodies and gepants; and to critically analyse the results of different studies and the profile of patients eligible for treatment with monoclonal antibodies, and the impact in terms of pharmacoeconomics.
ConclusionsThe clinical development of gepants, which are CGRP antagonists, for the acute treatment of migraine attacks, and CGRP ligand and receptor monoclonal antibodies offer promising results for these patients.
En los últimos años, una de las principales líneas de investigación en migraña se ha centrado en la búsqueda de marcadores biológicos que nos ayuden a entender mejor la fisiopatología de esta entidad y a diseñar nuevas opciones terapéuticas, inaugurando lo que se ha denominado «medicina de precisión en migraña». Moléculas como el péptido relacionado con el gen de la calcitonina (CGRP) y, en menor grado, el péptido activador de la adenilato-ciclasa pituitaria (PACAP) se había observado que estaban elevadas durante los ataques de migraña1 y en la migraña crónica (MC) tanto en líquido cefalorraquídeo2 como en suero3 y que su reducción farmacológica tenía una significación clínica con una mejoría en la migraña de los pacientes4,5. El desarrollo clínico de los gepantes, antagonistas del CGRP, para el tratamiento de los ataques de migraña se vio interrumpido temporalmente debido a una transaminitis6, si bien en el momento actual los resultados son prometedores7. Posteriormente, se desarrollaron anticuerpos monoclonales (AMC) contra ligando y contra el receptor del CGRP, sin interacción con las enzimas hepáticas, de gran tamaño (se evita así el paso a través de la barrera hematoencefálica [BHE]) y con elevada especificidad. Los ensayos clínicos con AMC contra el CGRP en migraña episódica (ME) y crónica y con los gepantes en el tratamiento del ataque han finalizado recientemente y ofrecen resultados esperanzadores para nuestros pacientes.
Desde el Grupo de Estudio de Cefaleas de la Sociedad Española de Neurología (GECSEN) nos planteamos la redacción de este documento, cuyo objetivo es abordar, basándonos en la evidencia publicada, cuestiones tan importantes como el papel del CGRP en la fisiopatología de la migraña, el mecanismo de acción de los AMC y de los gepantes, el análisis crítico de los resultados de los diferentes estudios, el perfil del paciente que podría ser candidato al tratamiento con AMC y su impacto en términos de farmacoeconomía.
MetodologíaSe creó un comité de expertos compuesto por 16 miembros del GECSEN que emitió de forma independiente sus recomendaciones para una serie de cuestiones, planteadas en formato pregunta-respuesta, de elevado interés en relación con los nuevos tratamientos que actúan sobre el CGRP. Estas recomendaciones se basan en la evidencia disponible y en la experiencia clínica. El método Delphi se utilizó para llegar a un consenso.
Siguiendo esta metodología se desglosaron los temas de mayor relevancia clínica a partir de las siguientes preguntas:
- 1.
¿Cuál es el papel del CGRP en la fisiopatología de la migraña?
- 2.
CGRP: ¿biomarcador de la migraña?
- 3.
Los gepantes en el tratamiento sintomático de la migraña
- 4.
¿Qué anticuerpos monoclonales disponen de ensayos clínicos en migraña? ¿Cuáles son los datos de eficacia y seguridad?
- 5.
¿Qué aportan los anticuerpos monoclonales frente a los tratamientos actuales en migraña episódica y crónica?
- 6.
¿Cuál sería el perfil del paciente respondedor a los anticuerpos monoclonales contra el péptido relacionado con el gen de la calcitonina?
- 7.
¿Cuándo se considera que la migraña es resistente al tratamiento?
- 8.
En términos de farmacoeconomía, ¿qué aportarán los anticuerpos monoclonales en el tratamiento de los pacientes con migraña?
En la última década, a raíz de los avances en investigación básica y de los diferentes estudios con neuroimagen funcional, conocemos más detalles acerca de la fisiopatología de una entidad tan compleja como es la migraña.
El protagonista principal en la génesis del dolor en el ataque de migraña es el sistema trigeminovascular (STV). Está constituido por los vasos meníngeos (durales y piales) y los aferentes sensitivos procedentes de la rama oftálmica del nervio trigémino que rodean dichos vasos (fibras nociceptoras polimodales Aδ y C). Estas fibras transmiten información nociceptiva al núcleo caudado del trigémino, donde forma parte del complejo trigeminocervical y recibe aferentes de las primeras raíces cervicales.
Existen 2 teorías no necesariamente excluyentes entre sí que explican por qué se activa el STV durante el ataque de migraña. La depresión cortical propagada, sustrato fisiopatológico del aura, es una onda de despolarización neuronal y glial que progresa lentamente a lo largo de la corteza cerebral a una velocidad de 2,5-5mm/seg y se sigue de una supresión sostenida de la actividad neuronal espontánea. Se acompaña de cambios en el calibre vascular y el flujo sanguíneo (una fase inicial de hiperemia cortical de pocos minutos de duración seguida de una fase de hipoperfusión más prolongada) y cambios en el metabolismo energético que implican la liberación de mediadores químicos al espacio extra- y perivascular (prostaglandinas y neurotransmisores excitatorios como glutamato y óxido nítrico entre otros). Durante este proceso se producen también cambios corticales como liberación de adenosín-trifosfato y glutamato por parte de la neurona y glía, así como activación de metaloproteasas que rompen la BHE y permiten que los mediadores químicos activen las terminales del trigémino que rodean los vasos meníngeos8.
Los núcleos del troncoencéfalo, locus coeruleus y núcleo dorsal del rafe, actuarían como generadores del dolor. Esta teoría se fundamenta en estudios de neuroimagen que han demostrado que estas estructuras se activan en esta fase de la migraña, si bien también lo hacen aquellas que integran el sistema límbico (córtex prefrontal, cíngulo, ínsula, tálamo). Además, estos núcleos, sobre todo la sustancia gris periacueductal, forman parte del sistema de control antinociceptivo descendente y, por lo tanto, pueden modular la señal de dolor. Su activación se traduce en una disfunción y crearía un ambiente permisivo para la entrada de aferencias nociceptivas9.
En ambos casos, la activación del STV es el resultado final. Se produce de forma bidireccional y tiene 2 consecuencias: una conducción ortodrómica, que transmite información nociceptiva hacia el núcleo caudal del trigémino, tálamo y córtex somatosensitiva, y una conducción antidrómica que genera una inflamación meníngea estéril, a partir de la liberación de neuropéptidos vasoactivos (sustancia P, CGRP, VIP, endotelina-3, PACAP, neuropéptido Y, péptido histidina metionina y neuroquinina A).
El CGRP es la molécula que se ha implicado con mayor consistencia en la activación del STV. Es uno de los 6 péptidos de la familia de las calcitoninas que en su subforma α es muy abundante en los vasos pericerebrales y en el ganglio de Gasser. Tiene una potente acción vasodilatadora y facilitadora de la nocicepción. Sabemos además que el CGRP y su receptor se expresan en neuronas distintas del ganglio de Gasser, como las terminaciones aferentes del núcleo caudal del trigémino (por lo que también interviene en el fenómeno de sensibilización central), el cerebelo, la duramadre, sustancia gris periacueductal, tálamo, hipotálamo, sistema límbico y córtex. Existe además una amplia evidencia sobre su implicación en el fenómeno de sensibilización central y, por ello, en la alodinia e hiperalgesia que experimentan algunos pacientes durante el ataque de migraña10–14. Finalmente, se ha descrito el papel del CGRP en la hipersensibilidad a la luz, uno de los síntomas más característicos, junto con el dolor, del ataque de migraña.
CGRP: ¿biomarcador de la migraña?Tal vez deberíamos comenzar preguntándonos si necesitamos un biomarcador para una entidad como la migraña, de diagnóstico clínico aparentemente sencillo. La respuesta a esta pregunta es definitivamente sí, y para ello daremos 2 posibles argumentos. En primer lugar, sabemos que existen errores diagnósticos, sobre todo en pacientes con MC15. En segundo lugar, el disponer de un biomarcador fiable nos permitiría evaluar la respuesta al tratamiento de forma objetiva. Además, en la migraña, aparentemente invisible, le da entidad biológica, algo que ayuda a reducir el estigma.
El dolor en un ataque de migraña se produce por la activación del STV, compuesto por un brazo aferente a cargo del nervio trigémino y un brazo eferente a cargo de la porción parasimpática del nervio facial. Parecería lógico buscar como potencial biomarcador de la migraña a alguno de los más de 10 neuropéptidos involucrados en la activación de dicho sistema, a nivel trigeminal (CGRP, sustancia P, neuroquinina A, amilina, colecistoquinina-8, S100B) y en el sistema parasimpático (VIP, PACAP, heliodermina, helospectina i y ii, péptido histidina-metionina, óxido nítrico).
En 1994, Lars Edvinsson y Peter Goadsby1 fueron capaces de medir varios de estos péptidos en muestras de sangre obtenidas en la vena yugular externa, ipsilateral al dolor, en el seno de una crisis de migraña. El único péptido que estaba elevado de forma consistente fue el CGRP. Aún más, el CGRP se normalizaba tras realizar un tratamiento adecuado con sumatriptán16 o, como más recientemente se ha demostrado, tras tratar la crisis con rizatriptán17. Sin embargo, los niveles de sustancia P, que entonces se pensaba era crucial, no se incrementaron y el VIP solo se elevó en algunos pacientes con síntomas autonómicos parasimpáticos. El problema para la reproducibilidad de estos trabajos era que se necesitaba hacer las mediciones en sangre yugular. Sin embargo, en el año 2012, Rodríguez-Osorio et al. demostraron, mediante técnicas de ELISA, que el CGRP también se elevaba en sangre periférica durante los ataques de migraña18. La importancia del CGRP quedó nuevamente de manifiesto cuando, en estudios experimentales, la inyección intravenosa de este neuropéptido fue capaz de inducir una cefalea tardía con características migrañosas solo a aquellos que tenían un diagnóstico de migraña19. Estos datos confirman al CGRP como un potencial biomarcador de la fase aguda de la migraña.
Posteriormente se demostró, en modelos animales, que el CGRP no solo estaba implicado en la fase de dolor agudo de la migraña, sino que también desempeñaba un papel relevante en el fenómeno de sensibilización y en la cronificación de la migraña20. No es sorprendente, por ello, que los niveles de CGRP se encuentren elevados en sangre periférica de pacientes con MC entre los ataques de migraña3. Otros estudios han demostrado que los niveles de CGRP están también aumentados en saliva o líquido cefalorraquídeo en el seno de una crisis de migraña en la ME, así como en la fase interictal de la MC2. No obstante, estos resultados han sido criticados por problemas metodológicos en el procesado de las muestras.
Un biomarcador debería ser capaz de predecir la respuesta al tratamiento específico y normalizarse con el tratamiento (piénsese en la glucemia para la diabetes). Ambos requisitos han sido demostrados razonablemente bien para el caso del CGRP y la MC: se ha comprobado que los niveles altos de este péptido pueden servir para predecir una buena respuesta terapéutica a la onabotulinumtoxinA (OnabotA) y que el CGRP desciende tras realizar un tratamiento adecuado con esta toxina pericranealmente4,5,21.
Con este cúmulo de datos podría pensarse que el CGRP en sangre periférica es el primer biomarcador de la migraña, tanto para la fase aguda como para su cronificación. Por desgracia, estamos aún lejos de poder afirmarlo categóricamente. Aproximadamente un tercio de los pacientes con migraña tienen niveles de CGRP similares a los de sujetos sin cefalea, tanto si lo determinamos en la fase aguda como en la fase interictal de la MC. Este es un punto crucial porque indicaría que para algunos pacientes con migraña el CGRP no sería el mediador más importante. De hecho, y ya lo vieron Edvinsson y Goadsby1, sabemos que también determinados péptidos parasimpáticos como el VIP están elevados en la crisis de migraña y en la MC22. Esto explicaría por qué los resultados de los gepantes para la fase aguda o de los anticuerpos frente al CGRP para el tratamiento sintomático no han sido tan espectaculares como uno esperaría si el CGRP fuera la única molécula implicada. Por otra parte, y aunque algunos datos experimentales lo apoyan23, una duda no resuelta es si el CGRP que se mide en la vena cubital procede realmente de la activación trigeminal. Otro problema importante es la reproducibilidad y fiabilidad de los test de ELISA existentes en el mercado. Por último, desconocemos la influencia de la corta vida media de estos péptidos en los resultados obtenidos en los diferentes laboratorios.
En síntesis, es cierto que necesitamos un biomarcador para la migraña accesible en sangre periférica y que el CGRP es el mejor posicionado. Sin embargo, estamos lejos de poder afirmar que el CGRP sea un marcador fiable y reproducible de migraña en nuestra práctica clínica asistencial.
Los gepantes en el tratamiento sintomático de la migrañaLos gepantes, antagonistas del CGRP, constituyen una nueva clase terapéutica para el tratamiento sintomático de la migraña, y una alternativa a los triptanes, agonistas de los receptores 5HT1B/1D24,25. En estudios aleatorizados, doble ciego y controlados con placebo, han demostrado ser más eficaces que placebo en la disminución de la intensidad del dolor, de forma similar a los triptanes, pero con un mejor perfil de seguridad.
Olcegepant (BIBN 4096BS) fue el primer gepante que demostró su eficacia frente a placebo en la disminución de la intensidad del dolor, con resultados prometedores en la tasa de recurrencia a las 2h, en la respuesta mantenida en las siguientes 24h y en la mejoría de síntomas asociados como las náuseas, fotofobia y sonofobia. El efecto adverso más frecuente fueron las parestesias26,27. Se administraba de forma de intravenosa, algo que no lo hacía práctico.
El desarrollo de telcagepant (MK-0974) se vio interrumpido, a pesar de sus buenos resultados frente a rizatriptán 10mg en un ensayo clínico aleatorizado, doble ciego y controlado con placebo28. La razón fue que, en un estudio posterior, que valoraba la administración diaria de telcagepant frente a placebo durante 3 meses como tratamiento preventivo en migraña, se describieron varios casos de toxicidad hepática6. Rimegepant (BMS-927711) ha demostrado ser eficaz frente a placebo en parámetros como ausencia de dolor a las 2h en un ensayo fase 2, con cifras comparables a las de sumatriptán 100mg. Actualmente se está llevando a cabo un ensayo fase 3 (NCT03235479)29.
Finalmente, ubrogepant (MK-1602) en ensayos fase 2 ha demostrado ser también más eficaz que placebo con una baja tasa de efectos adversos. Han finalizado recientemente 2 ensayos en fase 3 (NCT02828020 y NCT02867709) y permanece activa la fase de extensión abierta para valorar la tolerabilidad y seguridad del fármaco (NCT02873221)7.
La eficacia de los gepantes parece ser similar a la de los triptanes más potentes. Sus ventajas residen en la mayor duración de su efecto (que se traduciría en una ventaja farmacoeconómica al necesitar menos dosis), su mejor tolerabilidad, y, en concreto, la previsible ausencia de efectos secundarios cardiovasculares. Su principal desventaja frente alguno de los triptanes sería el paso de la BHE, generando algunos efectos adversos centrales (somnolencia).
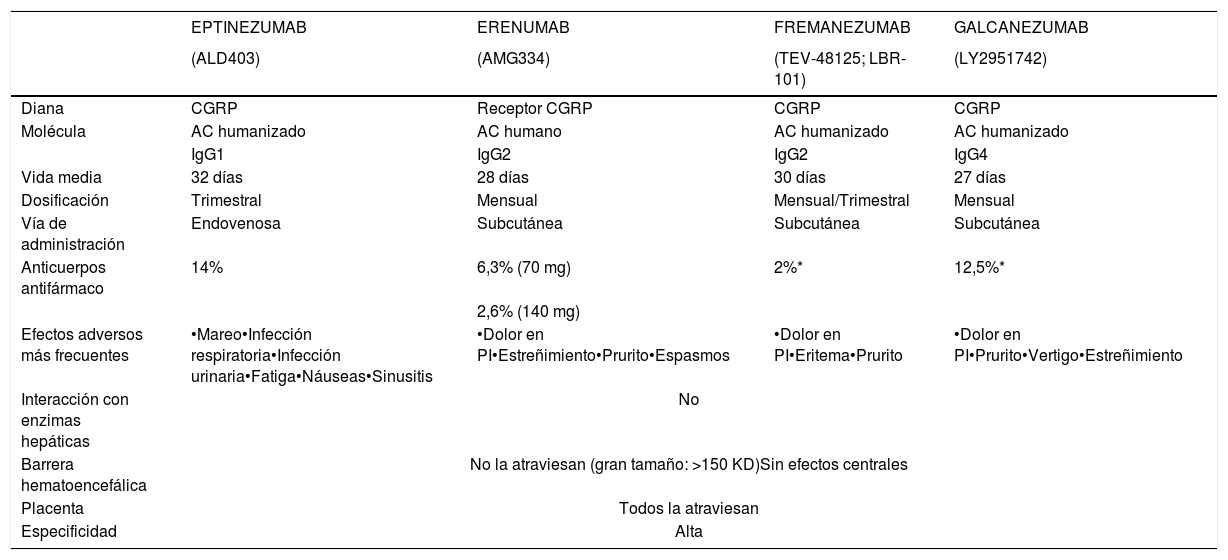
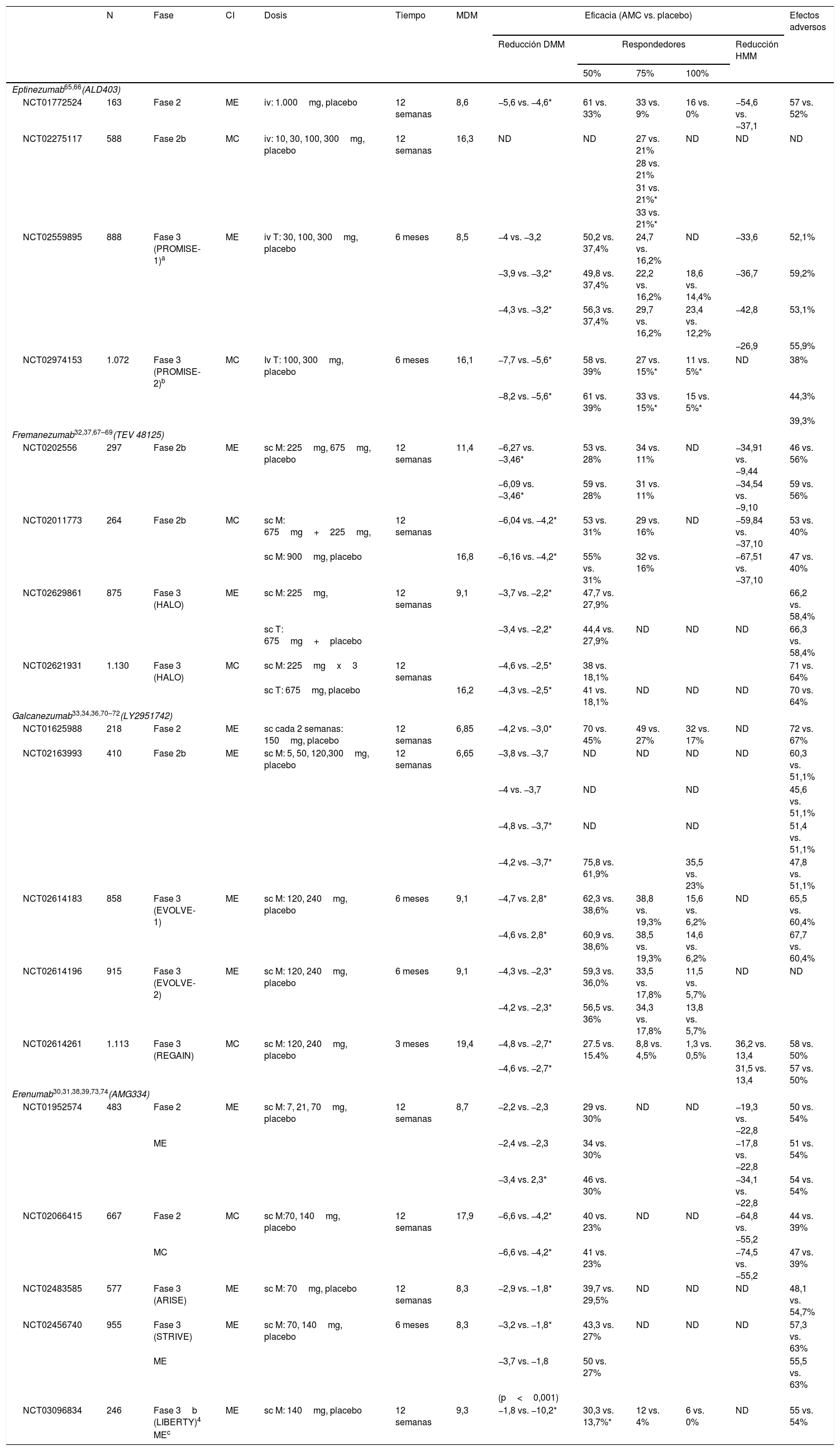
¿Qué anticuerpos monoclonales disponen de ensayos clínicos en migraña? ¿Cuáles son los datos de eficacia y seguridad?Los AMC en fase de desarrollo clínico son 4 en la actualidad: erenumab, fremanezumab, galcanezumab y eptinezumab. Sus principales características se resumen en la tabla 1. Hasta la fecha, se han publicado 8 ensayos clínicos en fase 3 en ME y MC (ver tabla 2), pero hay más ensayos en curso cuyos resultados conoceremos próximamente.
Características de los anticuerpos monoclonales
| EPTINEZUMAB | ERENUMAB | FREMANEZUMAB | GALCANEZUMAB | |
|---|---|---|---|---|
| (ALD403) | (AMG334) | (TEV-48125; LBR-101) | (LY2951742) | |
| Diana | CGRP | Receptor CGRP | CGRP | CGRP |
| Molécula | AC humanizado | AC humano | AC humanizado | AC humanizado |
| IgG1 | IgG2 | IgG2 | IgG4 | |
| Vida media | 32 días | 28 días | 30 días | 27 días |
| Dosificación | Trimestral | Mensual | Mensual/Trimestral | Mensual |
| Vía de administración | Endovenosa | Subcutánea | Subcutánea | Subcutánea |
| Anticuerpos antifármaco | 14% | 6,3% (70 mg) | 2%* | 12,5%* |
| 2,6% (140 mg) | ||||
| Efectos adversos más frecuentes | •Mareo•Infección respiratoria•Infección urinaria•Fatiga•Náuseas•Sinusitis | •Dolor en PI•Estreñimiento•Prurito•Espasmos | •Dolor en PI•Eritema•Prurito | •Dolor en PI•Prurito•Vertigo•Estreñimiento |
| Interacción con enzimas hepáticas | No | |||
| Barrera hematoencefálica | No la atraviesan (gran tamaño: >150 KD)Sin efectos centrales | |||
| Placenta | Todos la atraviesan | |||
| Especificidad | Alta | |||
Ensayos clínicos en migraña con anticuerpos monoclonales
| N | Fase | CI | Dosis | Tiempo | MDM | Eficacia (AMC vs. placebo) | Efectos adversos | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reducción DMM | Respondedores | Reducción HMM | ||||||||||
| 50% | 75% | 100% | ||||||||||
| Eptinezumab65,66(ALD403) | ||||||||||||
| NCT01772524 | 163 | Fase 2 | ME | iv: 1.000mg, placebo | 12 semanas | 8,6 | −5,6 vs. −4,6* | 61 vs. 33% | 33 vs. 9% | 16 vs. 0% | −54,6 vs. −37,1 | 57 vs. 52% |
| NCT02275117 | 588 | Fase 2b | MC | iv: 10, 30, 100, 300mg, placebo | 12 semanas | 16,3 | ND | ND | 27 vs. 21% | ND | ND | ND |
| 28 vs. 21% | ||||||||||||
| 31 vs. 21%* | ||||||||||||
| 33 vs. 21%* | ||||||||||||
| NCT02559895 | 888 | Fase 3 (PROMISE-1)a | ME | iv T: 30, 100, 300mg, placebo | 6 meses | 8,5 | −4 vs. −3,2 | 50,2 vs. 37,4% | 24,7 vs. 16,2% | ND | −33,6 | 52,1% |
| −3,9 vs. −3,2* | 49,8 vs. 37,4% | 22,2 vs. 16,2% | 18,6 vs. 14,4% | −36,7 | 59,2% | |||||||
| −4,3 vs. −3,2* | 56,3 vs. 37,4% | 29,7 vs. 16,2% | 23,4 vs. 12,2% | −42,8 | 53,1% | |||||||
| −26,9 | 55,9% | |||||||||||
| NCT02974153 | 1.072 | Fase 3 (PROMISE-2)b | MC | Iv T: 100, 300mg, placebo | 6 meses | 16,1 | −7,7 vs. −5,6* | 58 vs. 39% | 27 vs. 15%* | 11 vs. 5%* | ND | 38% |
| −8,2 vs. −5,6* | 61 vs. 39% | 33 vs. 15%* | 15 vs. 5%* | 44,3% | ||||||||
| 39,3% | ||||||||||||
| Fremanezumab32,37,67–69(TEV 48125) | ||||||||||||
| NCT0202556 | 297 | Fase 2b | ME | sc M: 225mg, 675mg, placebo | 12 semanas | 11,4 | −6,27 vs. −3,46* | 53 vs. 28% | 34 vs. 11% | ND | −34,91 vs. −9,44 | 46 vs. 56% |
| −6,09 vs. −3,46* | 59 vs. 28% | 31 vs. 11% | −34,54 vs. −9,10 | 59 vs. 56% | ||||||||
| NCT02011773 | 264 | Fase 2b | MC | sc M: 675mg+225mg, | 12 semanas | −6,04 vs. −4,2* | 53 vs. 31% | 29 vs. 16% | ND | −59,84 vs. −37,10 | 53 vs. 40% | |
| sc M: 900mg, placebo | 16,8 | −6,16 vs. −4,2* | 55% vs. 31% | 32 vs. 16% | −67,51 vs. −37,10 | 47 vs. 40% | ||||||
| NCT02629861 | 875 | Fase 3 (HALO) | ME | sc M: 225mg, | 12 semanas | 9,1 | −3,7 vs. −2,2* | 47,7 vs. 27,9% | 66,2 vs. 58,4% | |||
| sc T: 675mg+placebo | −3,4 vs. −2,2* | 44,4 vs. 27,9% | ND | ND | ND | 66,3 vs. 58,4% | ||||||
| NCT02621931 | 1.130 | Fase 3 (HALO) | MC | sc M: 225mgx3 | 12 semanas | −4,6 vs. −2,5* | 38 vs. 18,1% | 71 vs. 64% | ||||
| sc T: 675mg, placebo | 16,2 | −4,3 vs. −2,5* | 41 vs. 18,1% | ND | ND | ND | 70 vs. 64% | |||||
| Galcanezumab33,34,36,70–72(LY2951742) | ||||||||||||
| NCT01625988 | 218 | Fase 2 | ME | sc cada 2 semanas: 150mg, placebo | 12 semanas | 6,85 | −4,2 vs. −3,0* | 70 vs. 45% | 49 vs. 27% | 32 vs. 17% | ND | 72 vs. 67% |
| NCT02163993 | 410 | Fase 2b | ME | sc M: 5, 50, 120,300mg, placebo | 12 semanas | 6,65 | −3,8 vs. −3,7 | ND | ND | ND | ND | 60,3 vs. 51,1% |
| −4 vs. −3,7 | ND | ND | 45,6 vs. 51,1% | |||||||||
| −4,8 vs. −3,7* | ND | ND | 51,4 vs. 51,1% | |||||||||
| −4,2 vs. −3,7* | 75,8 vs. 61,9% | 35,5 vs. 23% | 47,8 vs. 51,1% | |||||||||
| NCT02614183 | 858 | Fase 3 (EVOLVE-1) | ME | sc M: 120, 240mg, placebo | 6 meses | 9,1 | −4,7 vs. 2,8* | 62,3 vs. 38,6% | 38,8 vs. 19,3% | 15,6 vs. 6,2% | ND | 65,5 vs. 60,4% |
| −4,6 vs. 2,8* | 60,9 vs. 38,6% | 38,5 vs. 19,3% | 14,6 vs. 6,2% | 67,7 vs. 60,4% | ||||||||
| NCT02614196 | 915 | Fase 3 (EVOLVE-2) | ME | sc M: 120, 240mg, placebo | 6 meses | 9,1 | −4,3 vs. −2,3* | 59,3 vs. 36,0% | 33,5 vs. 17,8% | 11,5 vs. 5,7% | ND | ND |
| −4,2 vs. −2,3* | 56,5 vs. 36% | 34,3 vs. 17,8% | 13,8 vs. 5,7% | |||||||||
| NCT02614261 | 1.113 | Fase 3 (REGAIN) | MC | sc M: 120, 240mg, placebo | 3 meses | 19,4 | −4,8 vs. −2,7* | 27.5 vs. 15.4% | 8,8 vs. 4,5% | 1,3 vs. 0,5% | 36,2 vs. 13,4 | 58 vs. 50% |
| −4,6 vs. −2,7* | 31,5 vs. 13,4 | 57 vs. 50% | ||||||||||
| Erenumab30,31,38,39,73,74(AMG334) | ||||||||||||
| NCT01952574 | 483 | Fase 2 | ME | sc M: 7, 21, 70mg, placebo | 12 semanas | 8,7 | −2,2 vs. −2,3 | 29 vs. 30% | ND | ND | −19,3 vs. −22,8 | 50 vs. 54% |
| ME | −2,4 vs. −2,3 | 34 vs. 30% | −17,8 vs. −22,8 | 51 vs. 54% | ||||||||
| −3,4 vs. 2,3* | 46 vs. 30% | −34,1 vs. −22,8 | 54 vs. 54% | |||||||||
| NCT02066415 | 667 | Fase 2 | MC | sc M:70, 140mg, placebo | 12 semanas | 17,9 | −6,6 vs. −4,2* | 40 vs. 23% | ND | ND | −64,8 vs. −55,2 | 44 vs. 39% |
| MC | −6,6 vs. −4,2* | 41 vs. 23% | −74,5 vs. −55,2 | 47 vs. 39% | ||||||||
| NCT02483585 | 577 | Fase 3 (ARISE) | ME | sc M: 70mg, placebo | 12 semanas | 8,3 | −2,9 vs. −1,8* | 39,7 vs. 29,5% | ND | ND | ND | 48,1 vs. 54,7% |
| NCT02456740 | 955 | Fase 3 (STRIVE) | ME | sc M: 70, 140mg, placebo | 6 meses | 8,3 | −3,2 vs. −1,8* | 43,3 vs. 27% | ND | ND | ND | 57,3 vs. 63% |
| ME | −3,7 vs. −1,8 | 50 vs. 27% | 55,5 vs. 63% | |||||||||
| (p<0,001) | ||||||||||||
| NCT03096834 | 246 | Fase 3b (LIBERTY)4 | ME | sc M: 140mg, placebo | 12 semanas | 9,3 | −1,8 vs. −10,2* | 30,3 vs. 13,7%* | 12 vs. 4% | 6 vs. 0% | ND | 55 vs. 54% |
| MEc | ||||||||||||
AMC: anticuerpo monoclonal; CGRP: péptido relacionado con el gen de la calcitonina; CI: criterios de inclusión; DMM: días de migraña al mes; HMM: horas de migraña al mes; iv: intravenoso; M: mensual; MC: migraña crónica; MDM: media días de migraña al mes; ME: migraña episódica; N: tamaño muestral; ND: no disponible; sc: subcutáneo, T: trimestral.
En ME el diseño de los ensayos fase 3 con los diferentes AMC es similar: multicéntrico, aleatorizado, doble ciego, controlado con placebo y 3 brazos, 2 de ellos con dosis diferente del fármaco. La variable principal es el número de días de migraña al mes y se considera que el paciente es respondedor si esta cifra se reduce como mínimo un 50% tras 12 semanas de terapia. La media de días de migraña/mes fue de 8,3-9,1 en el período basal en los diferentes ensayos. Todos los AMC han demostrado ser eficaces frente a placebo en la prevención de la migraña. A continuación enumeramos los ensayos fase 3 finalizados con resultados comunicados:
ErenumabEnsayo STRIVE (NCT02456740). Incluyó a 955 pacientes distribuidos en 3 brazos tratados durante 24 semanas: 317 (70mg/mes de erenumab), 319 (140mg/mes de erenumab) y 319 pacientes tratados con placebo. Ambas dosis de erenumab fueron superiores frente al placebo en la disminución del número de días de migraña y en las variables secundarias: días de migraña con necesidad de tratamiento agudo, escalas de discapacidad y escalas de actividades de la vida diaria30. Se permitió la inclusión de pacientes con tratamiento preventivo oral en monoterapia a dosis estable.
Ensayo ARISE (NCT02483585). Incluyó a 577 pacientes que recibieron 70mg/mes de erenumab (282 pacientes) versus placebo (288 pacientes) durante 12 semanas. Erenumab logró una reducción superior al placebo en días de migraña al mes y en las variables secundarias (las mismas del ensayo STRIVE)31.
FremanezumabEnsayo NCT02629861. Incluyó a 875 pacientes en 3 brazos: 290 pacientes (225mg de fremanezumab mensual), 291 pacientes (675mg de fremanezumab trimestral) y 294 pacientes (placebo). Fremanezumab redujo el número de días de migraña/mes, el número de días de migraña con necesidad de tratamiento agudo y la puntuación en la escala Migraine Disability Assessment (MIDAS) de forma significativa frente a placebo32. Se permitió la inclusión de pacientes con tratamiento preventivo oral en monoterapia a dosis estable.
GalcanezumabEnsayo EVOLVE1 (NCT02614183) y EVOLVE2 (NCT02614196). Incluyeron respectivamente a 858 y a 915 pacientes tratados durante 6 meses distribuidos en 3 brazos: galcanezumab 120mg, galcanezumab 240mg y placebo. Ambas dosis de galcanezumab fueron superiores frente a placebo en la variable primaria y en escalas de calidad de vida33,34.
EptinezumabEnsayo NCT02974153 (PROMISE1). Incluyó a 888 pacientes en 4 brazos tratados con 300, 100, 30mg de eptinezumab y placebo. Las 3 dosis de eptinezumab disminuyeron de forma significativa frente a placebo el número días de migraña al mes y consiguieron una mayor proporción de pacientes con reducción del número de crisis (≥50%)35.
Eficacia en migraña crónicaActualmente, solo fremanezumab y galcanezumab han publicado resultados en ensayos fase 3. No se dispone de información sobre el ensayo NCT02559895 (PROMISE2) de eptinezumab. Erenumab ha demostrado ser eficaz en ensayos fase 2 y tiene estudios abiertos de seguimiento pero no ha realizado ensayos fase 3 en MC.
Ensayo NCT02614261 (REGAIN)36. Este estudio con galcanezumab demostró una reducción media de los días de migraña al mes de 4,3 días con la dosis de 120mg (n=226) y de 4,6 días con la de 240mg (n=220) frente al grupo placebo (n=450).
Ensayo NCT02621931. Incluyó a 1.130 pacientes distribuidos en 3 brazos con un seguimiento de 16 semanas: 675mg de fremanezumab (376 pacientes), 225mg (379 pacientes) y 375 pacientes tratados con placebo. La disminución en el número de días de cefalea al mes fue superior para fremanezumab así como la respuesta de las variables secundarias (días de migraña al mes y puntuación en la escala Headache Impact Test (HIT-6])37. Se permitió la inclusión de pacientes con tratamiento preventivo oral en monoterapia a dosis estable.
Eficacia en migraña refractariaSe han planteado estudios en pacientes con migraña que no han respondido a 2-4 clases de tratamientos con fremanezumab (FOCUS), erenumab (LIBERTY) y galcanezumab (CONQUER). Hasta ahora se han publicado resultados con erenumab en pacientes con ME (estudio LIBERTY–NCT03096834) donde erenumab 140mg (n=121) consiguió una reducción del 50% o más de los días mensuales de migraña en un 30% de los pacientes frente a un 14% en el grupo placebo (n=125)38 y un subanálisis de pacientes con erenumab en MC39 que fallaron a≥1 o≥2 tratamientos previos.
Datos de seguridadA diferencia de lo que ocurre con los gepantes, el metabolismo de los AMC no es hepático, y, dado que no atraviesan la BHE, no se esperan efectos adversos a nivel del sistema nervioso central, salvo en aquellas estructuras no protegidas por esta barrera, como la glándula hipofisaria. Sin embargo, se ha planteado que el bloqueo mantenido del CGRP pueda suponer un riesgo a diferentes niveles en los que el papel de este neuropéptido, en animales de experimentación es clave: piel (cicatrización de las heridas), sistema gastrointestinal (protector mucoso y de motilidad) y cardiovascular (protector de la isquemia cardiaca y cerebral y frente a la hipertensión), metabolismo óseo y filtrado glomerular
Hasta ahora la tasa de EA en los grupos activos y placebo es muy similar y no se han detectado EA graves, si bien la duración de los ensayos es relativamente corta para una patología crónica como la migraña. Cabe destacar de forma global el dolor en el punto de inyección, con diferencias escasas o ausentes frente a placebo, la induración o eritema con fremanezumab y de prurito o reacción local con galcanezumab. La frecuencia de infección respiratoria, urinaria, fatiga o náuseas no muestra diferencias significativas frente a placebo y no se han registrado problemas vasculares. La tasa de abandono en todos los ensayos ha resultado baja y por EA muy baja, siempre<4%, en contraste con las cifras registradas para los TPO40. Sin embargo, dado que no podemos descartar EA a largo plazo, deberán garantizarse registros en este sentido para la obtención de más datos de seguridad.
¿Qué aportan los anticuerpos monoclonales frente a los tratamientos actuales en migraña episódica y crónica?En términos comparativos con las opciones de tratamiento disponibles para la prevención de la migraña en el momento actual, los AMC suponen un hito, ya que, hasta la fecha, todas las herramientas terapéuticas eran heredadas de otras ramas del conocimiento médico (antihipertensivos, antidepresivos, neuromoduladores entre otros). Los nuevos AMC han tenido un desarrollo clínico similar al de otros anticuerpos para otras patologías y las evidencias actuales indican un efecto potencialmente beneficioso tanto en ME como en MC.
Se considera que todos los pacientes con MC y hasta el 40% de los que sufren ME podrían ser candidatos a iniciar un tratamiento preventivo que intente reducir el impacto de la enfermedad y la discapacidad que produce, aunque solo un porcentaje muy bajo acaba accediendo a los mismos41. Las terapias actualmente disponibles presentan varios problemas que dificultan su correcta aplicación en términos de eficacia, tolerabilidad, adherencia o interacciones. Aproximadamente uno de cada 5 pacientes abandona el tratamiento por los EA42 y solo uno de cada 5 lo mantiene de forma adecuada cuando este se alarga más de un año43. Respecto a la MC, la forma más invalidante de la enfermedad, en Estados Unidos solamente la infiltración periódica con OnabotA ha sido aprobada específicamente para el tratamiento de esta entidad44.
En este sentido, podemos considerar que los nuevos AMC tendrán las siguientes diferencias y aportarán los siguientes puntos con respecto a los tratamientos clásicos:
- 1.
Vía de administración y pauta: con la excepción de OnabotA y los bloqueos anestésicos, la mayor parte de los preventivos disponibles se pautan por vía oral y diaria. Sin embargo, los AMC se administrarán por vía subcutánea (zona de deltoides, muslo o abdomen) y mensual o trimestral, salvo eptinezumab que será intravenoso y trimestral. Una diferencia importante consistirá en que, previsiblemente, este tipo de moléculas serán de dispensación en farmacias hospitalarias al igual que otros monoclonales, por lo que los centros sanitarios tendrán que articular mecanismos administrativos internos para tal fin. Es de esperar, por tanto, que al menos inicialmente estos tratamientos se reserven para casos refractarios y posiblemente requieran algún tipo de valoración específica en consultas especializadas de cefalea.
- 2.
Mecanismo de acción: aunque otros tratamientos para migraña han actuado sobre la vía del CGRP, como por ejemplo los triptanes, gepantes, metisergida o incluso OnabotA, hasta la fecha ningún tratamiento ha sido diseñado para establecer un bloqueo tan selectivo de dicha molécula.
- 3.
Adherencia: uno de los principales problemas que lastra en muchas ocasiones la eficacia de los tratamientos actuales es la falta de adherencia de los pacientes y el cumplimiento erróneo de las pautas terapéuticas dadas por los facultativos. Es de esperar que estos nuevos tratamientos consigan una adherencia mayor porque serán administrados en el propio hospital o con autoinyectores que conlleven una estricta educación de los pacientes.
- 4.
Eficacia y seguridad: este punto ya ha sido comentado ampliamente en otro apartado de esta revisión. Es pronto para determinar qué aspectos de eficacia podrán mostrar ventajas con respecto a otros tratamientos canónicos, dado que no existen estudios comparativos todavía45. Sí debe mencionarse que la respuesta a estos tratamientos se inicia de forma temprana, ya en los primeros días, a diferencia de lo que ocurre con los TPO. En términos de seguridad, los datos disponibles de los diversos ensayos clínicos son muy prometedores, tanto por la ausencia de eventos graves como por las bajas tasas de EA que, previsiblemente, también mejorarán aspectos relacionados con la adherencia. Los efectos secundarios sí son un problema frecuente en los tratamientos actuales.
- 5.
Interacciones y contraindicaciones: hasta la fecha no se han descrito interacciones relevantes con los AMC. Ello representa una gran ventaja ya que, en la migraña, por la comorbilidad asociada y el uso excesivo de medicación, los TPO pueden generar problemas de interacción, bien entre ellos, bien con otros grupos terapéuticos46. No se han definido contraindicaciones hasta el momento actual y debe evitarse su uso en el embarazo.
- 6.
Facilidad de la pauta terapéutica: los ensayos disponibles han sido realizados de forma sistemática en monoterapia y aportan datos de eficacia evidentes. Dado que una gran mayoría de pacientes, sobre todo los más refractarios, son usuarios de terapias complejas, es muy probable que en muchos casos se simplifiquen los esquemas terapéuticos, ya que además no se precisa titulación. También está por ver cómo será el uso de los monoclonales como terapia añadida o en pautas de politerapia y qué sinergias puedan ser las más racionales.
Es difícil precisar cuál sería el perfil del paciente respondedor a los AMC contra el receptor o ligando de CGRP. En los ensayos clínicos realizados hasta la fecha, estos fármacos han demostrado ser eficaces en contextos diversos: migraña con y sin aura, migraña episódica y crónica, migraña con y sin respuesta a tratamientos preventivos previos, o incluso sin exposición previa a tratamientos preventivos, migraña con o sin abuso de analgésicos, y migraña con o sin comorbilidad asociada. Como consecuencia de ello, disponemos ya de la aprobación de la Food and Drug Administration (FDA) en Estados Unidos para erenumab, fremanezumab y galcanezumab como tratamiento preventivo de la migraña sin ninguna restricción o limitación. En principio, cualquier paciente con 4 o más crisis al mes de migraña podría beneficiarse del tratamiento.
Si bien esto es así, y con los datos con los que contamos hasta la fecha, debemos admitir que la magnitud del efecto beneficioso no es la misma para todos los pacientes. A la luz de los ensayos publicados, da la sensación de que hay un grupo de hiperrespondedores, otros con nivel de respuesta medio y otros pacientes con escasa o nula respuesta47. El problema es que, hoy por hoy, no se ha publicado ningún factor predictor de respuesta, y solo podemos hipotetizar acerca de qué pacientes migrañosos van a responder mejor.
Dadas las características de los AMC, cabría considerar que el principal candidato para recibirlo, y en quien se esperaría logar un mayor efecto, sería el paciente con migraña con elevada frecuencia e intensidad de crisis de migraña, en el que se produciría una liberación recurrente y sostenida de CGRP. La determinación programada de niveles plasmáticos de este péptido durante las crisis y en período intercrítico de migraña apoya esta hipótesis20. Para corroborar esta afirmación sería de gran interés demostrar que aquellos pacientes con migraña con niveles plasmáticos elevados de CGRP tienen una mejor respuesta a los AMC y que, tras la administración de los mismos, disminuyen en los pacientes más respondedores. Queda por dilucidar si esta hipótesis es valorable para el erenumab (único AMC que actúa sobre el receptor del CGRP y no sobre el ligando del péptido), ya que es posible que las determinaciones plasmáticas de CGRP no tengan la misma significación que para los otros AMC. Desde un punto de vista teórico, aquellos pacientes con mayor preeminencia de la vía del CGRP serían los más respondedores a los AMC. Por el contrario, aquellos en los que otras vías tuvieran mayor protagonismo (como las del VIP, PACAP 38, u otros neuropéptidos inflamatorios relacionados con las crisis de migraña y su perpetuación o cronificación), deberían de tener una respuesta a claramente inferior.
¿Cuándo se considera que la migraña es resistente al tratamiento?La migraña refractaria implica per se una mayor discapacidad e impacto en la calidad de vida. Aun cuando se desconoce su prevalencia en la población general, en la serie de Irimia et al. representa el 5,1% de los pacientes atendidos de forma de forma consecutiva en una consulta monográfica de cefaleas48. No existe un consenso en cuanto a sus criterios diagnósticos49–56 y, pese a la recomendación de grupos de expertos, no forma parte de la tercera edición de la Clasificación Internacional de Cefaleas (CIC-3)57.
En la práctica clínica, utilizamos este término para describir a aquellos pacientes con migraña que combinan una importante discapacidad e impacto en la calidad de vida, y la ausencia de respuesta o intolerancia a varios tratamientos preventivos, pautados en monoterapia o en terapia combinada.
En el año 2008 la Refractory Headache Special Interest Section de la Sociedad Americana de Cefaleas (American Headache Society, AHS) propuso sus criterios de migraña refractaria, válidos tanto para la ME como para la MC50. La cefalea del paciente debía interferir en su calidad de vida a pesar de la modificación y optimización de los 3 pilares básicos en el manejo terapéutico de la migraña: hábitos y factores desencadenantes, tratamiento agudo sintomático y tratamiento preventivo. Se requería el fracaso de al menos 2 de los siguientes 4 grupos terapéuticos: betabloqueantes, neuromoduladores, antagonistas del calcio y antidepresivos tricíclicos, en monoterapia o terapia combinada. El tratamiento debía mantenerse al menos durante 2 meses a dosis óptima para considerar la ausencia de respuesta (en una encuesta posterior dirigida a los miembros de la AHS, alrededor del 40% consideraban más adecuado incrementar el número de tratamientos preventivos ensayados a 3 o, incluso, 4 fármacos54).
Los autores establecían también como necesario el fracaso de 2 posibles estrategias en el tratamiento sintomático de los ataques: triptanes o dihidroergotamina nasal o inyectable, o antiinflamatorios no esteroideos o analgésicos solos o en combinación. Al valorar, además, como criterio operativo la discapacidad secundaria del paciente (entendida como una puntuación en la escala MIDAS≥11, se introduce un nuevo concepto, la repercusión de la migraña en la calidad vida del paciente. Finalmente, se consideró aceptable la posible coexistencia de un uso excesivo de medicación.
En 2014, la Federación Europea de Cefaleas (European Headache Federation, EHF) propuso sus criterios de migraña refractaria, aplicables solo a la MC56. El diagnóstico de esta entidad se centra en el fracaso al menos de 3 grupos terapéuticos (se incluye OnabotA) que debían mantenerse durante un mínimo de 3 meses. Incide también en la necesidad de descartar la cefalea relacionada con el uso excesivo de medicación sintomática mediante un procedimiento de deshabituación, farmacológico (oral/intravenoso) o educacional.
Es difícil, en nuestra opinión, optar por una de ambas definiciones. La americana50 tiene la ventaja de considerar la discapacidad del paciente y la respuesta al tratamiento sintomático (aunque la dihidroergotamina intranasal no está disponible en Europa), y no requerir un proceso de deshabituación. Además, incluye la ME, cuya línea divisoria con la MC es con frecuencia permeable, como demuestran algunos estudios epidemiológicos58. En cuanto a la europea56, define con más claridad las pruebas necesarias para descartar una cefalea secundaria, y tiene en consideración el tratamiento preventivo con OnabotA.
Un análisis crítico de ambas propuestas nos lleva a plantear, en relación con la ME refractaria, la posibilidad de tratamiento con OnabotA según recomendación del GECSEN59,60, si los tratamientos preventivos fracasan o no son bien tolerados. Por otro lado, tanto en la definición de ME refractaria como en la de MC refractaria, sería razonable incluir el bloqueo anestésico del nervio occipital mayor como posible opción terapéutica (nivel de evidencia iv y nivel de evidencia iiib respectivamente)61. Tampoco parece adecuado que fármacos con el máximo nivel de evidencia en MC (topiramato y OnabotA) tengan el mismo peso que otros considerados de segunda línea en el manejo de esta entidad56.
En términos de farmacoeconomía ¿qué aportarán los anticuerpos monoclonales en el tratamiento de los pacientes con migraña?En el momento de escribir esta revisión no disponemos del precio y reembolso en España para los AMC, cuya comercialización representa un gran avance para el tratamiento preventivo de la migraña62. Una vez demostrada la eficacia y seguridad de estas terapias, el clínico debe disponer de información complementaria en términos de farmacoeconomía. La consideración de esta perspectiva en el tratamiento de la migraña tiene una gran importancia teniendo en cuenta la elevada prevalencia de la enfermedad, la limitación de los recursos económicos de los que dispone el sistema de salud y la existencia de diferentes alternativas terapéuticas eficaces ya comercializadas. Este tipo de estudios permiten establecer qué tratamiento es el más eficiente, es decir, el que obtiene mejores resultados clínicos con el menor coste. La evaluación económica de los fármacos preventivos de migraña debe tener en cuenta su impacto sobre los costes directos (gasto en consultas médicas, visitas a urgencias, precio del fármaco), indirectos (pérdida de productividad) y la calidad de vida del paciente.
En la actualidad, la información farmacoeconómica de los AMC es muy escasa y no existen estudios comparativos frente a TPO o infiltración de OnabotA, en términos de eficacia o coste. En publicaciones recientes63, erenumab ha demostrado que reduce el coste directo e indirecto asociado a la cefalea en pacientes con ME y MC (que habían utilizado previamente al menos una terapia preventiva) y aumenta los años de vida ajustados por calidad en comparación con la no utilización de tratamiento preventivo. Los cálculos de costes están hechos en función del sistema de salud americano y es importante subrayar que los resultados podrían ser incluso mejores de los que refieren los autores, ya que el estudio está hecho antes de la comercialización de erenumab y se asumen costes por encima del precio final de mercado63.
Por otro lado, el Instituto de Revisión Clínica y Económica estadounidense (ICER), un organismo de investigación independiente sin ánimo de lucro, publicó recientemente su informe final64 en el que se analiza la evidencia sobre la efectividad y si el precio de los AMC en migraña es apropiado en relación con los beneficios clínicos que ofrecen (valor a largo plazo por el coste del tratamiento). Se consideró que el valor a largo plazo de erenumab (único anticuerpo con precio público disponible) es intermedio en adultos con MC e intermedio-bajo en ME.
Es probable que en los próximos meses comiencen a aparecer publicaciones al respecto ya que la presentación de estudios de farmacoeconomía con los nuevos medicamentos que se registren es un requisito para la negociación del precio y reembolso en diferentes países europeos. Además, está previsto que a finales de 2018 se disponga de las guías del National Institute for Health and Care Excellence (NICE) en las que se analizará la efectividad clínica y coste de los diferentes AMC contra el CGRP dentro de los procedimientos para comercialización de estos fármacos en el Reino Unido.
Los AMC son fármacos seguros, eficaces y bien tolerados65. Es razonable pensar que la reducción del número de días de migraña y la discapacidad que esta genera se asocien a una disminución de los costes directos e indirectos y a una mejoría en la calidad de vida del paciente. La escasa evidencia disponible apoya esta afirmación. Sin embargo, son necesarios más estudios farmacoeconómicos que sustenten los beneficios que pueden obtenerse con estas terapias y que ayuden al clínico a elegir de forma racional entre las actuales alternativas terapéuticas, lo que redundará en un mejor uso de los recursos disponibles y en un incremento de la calidad asistencial.
FinanciaciónEste documento no ha recibido ninguna financiación.
Conflicto de interesesLos autores presentan los siguientes conflictos de interés:
El dr. Belvís ha recibido honorarios de Allergan, Chiesi, Novartis, Teva, Juste y Eli Lilly.
El dr. Guerrero Peral ha recibido honorarios de Allergan, Alder, Amgen, Chiesi, Eli Lilly, Novartis, y Teva.
El dr. Díaz Insa ha recibido honorarios de Allergan, Alter, Kern Pharma, Lundbeck, Novartis y Teva.
La dra. Gago Veiga ha recibido honorarios de Allergan, Eisai, Novartis, Teva y UCB Pharma.
La dra. Cuadrado ha recibido honorarios de Almirall, Allergan, Juste y Novartis.
El dr. Huerta ha recibido honorarios de Almirall, Allergan, Chiesi, Eisei, Kern Pharma, MSD, Eli Lilly, Teva y Zambón.
El dr. Irimia ha recibido honorarios de Alder, Allergan, Eli Lilly y Novartis.
El dr. Láinez ha recibido honorarios de Allergan, Amgen, ATI, Bial, Boehringher, Chiesi, Eisai, ElectroCore, Eli Lilly, Medtronic, Novartis, Otsuka, Roche, Teva y UCB.
El dr. Latorre ha recibido honorarios de Allergan, Chiesi, Novartis y Teva.
El dr. Leira ha recibido honorarios por ensayos clínicos, conferencias y asesoramiento de Allergan, Amgen, Chiesi, Eli Lilly, Novartis y Teva.
El dr. Pascual ha recibido honorarios de Almirall, Amgen, Novartis, Stendhal y Teva.
El dr. Porta-Etessam ha recibido honorarios de Allergan, Chiesi, Eisai, Exeltis, Grunenthal, Novartis y Teva.
La dra. Pozo-Rosich ha recibido honorarios de Allergan, Almirall, Amgen, Chiesi, Eli Lilly, Novartis y Teva.
La dra. Sánchez del Río ha recibido honorarios de Allergan, Chiesi, Eli Lilly, Novartis y Teva.
La dra. Santos Lasaosa ha recibido honorarios de Allergan, Amgen, Chiesi, Eisai, Exeltis, Novartis y Teva.
El dr. Viguera ha recibido honorarios de Allergan y Novartis.

