



Existe información limitada de la realización de diagnóstico presintomático en ataxias espinocerebelosas (SCA) autosómicas dominantes. La llegada del diagnóstico molecular, además de brindar la posibilidad de realizar identificación en pacientes portadores de distintas enfermedades, permitió también la posibilidad de detectar enfermedades incluso antes de su presentación. Esto atrajo la atención sobre las implicaciones éticas que deberían ser consideradas en estos sujetos, con la finalidad de salvaguardar su bienestar físico y psicológico.
DesarrolloLa SCA está compuesta por un grupo de trastornos neurodegenerativos con patrón de herencia autosómico dominante. Existen pocas publicaciones que describen el proceso de asesoramiento y los lineamientos considerados durante el proceso de diagnóstico presintomático. El número de integrantes de los equipos multidisciplinarios, sus áreas de especialidad y número de sesiones durante el asesoramiento es variable en cada uno de los trabajos analizados. Sin embargo, las bases para su realización tienen origen en documentos comunes, en los cuales algunos de los autores han participado en fechas más recientes.
ConclusionesEl diagnóstico presintomático debe ser realizado bajo lineamientos que salvaguarden el bienestar de los sujetos. Sería recomendable que el diagnóstico de SCA sea realizado solo a pacientes con clínica sugestiva, mayores de 18 años y con un riesgo mínimo del 50%. Deben estar disponibles esquemas de asesoramiento genético en todos aquellos centros que pretenden realizar diagnóstico de SCA antes de la presentación de síntomas.
Information on achieving presymptomatic diagnosis of spinocerebellar ataxia (SCA) is limited. The advent of molecular diagnosis makes it possible to identify the carriers of different diseases and has also introduced the prospect of detecting diseases even before their onset. This has drawn attention to the ethical implications that must be considered in these subjects with a view to preserving their physical and psychological well-being.
DevelopmentSCA is composed of a group of neurodegenerative disorders with autosomal dominant inheritance. Only a few publications have described the genetic counselling processes and guidelines to be followed during the process of presymptomatic diagnosis (PSD). The size of the multidisciplinary teams, their areas of expertise, and the number of counselling sessions are different for each of the studies analysed here. However, the basis of presymptomatic diagnosis originates in common guidelines to which members of our team have contributed recently.
ConclusionPresymptomatic diagnosis should be performed according to guidelines that safeguard the subjects’ welfare. The diagnostic process is only recommended for patients over 18 years old with symptoms suggesting SCA, and a minimum risk of 50%. Genetic counselling programmes must be available in all centres that offer presymptomatic diagnosis of SCA.
Con la llegada del diagnóstico molecular y más tarde programas como el Proyecto del Genoma Humano se esperaba el advenimiento de grandes beneficios en distintas áreas del conocimiento, así como la posibilidad de brindar ventajas para la salud. Los objetivos primarios de la medicina genética fueron claros: diagnóstico, tratamiento y prevención de trastornos genéticos1. Como resultado de esto, desde hace más de 3 décadas es posible realizar el diagnóstico molecular de trastornos genéticos como la enfermedad de Huntington (HD), Alzheimer, ataxias espinocerebelosas (SCA), polineuropatía familiar amiloide, entre otros trastornos neurodegenerativos. Lo anterior brindó la posibilidad de realizar los diagnósticos presintomático (DPS) y predictivo de algunas enfermedades neurodegenerativas antes de la presentación de signos y síntomas2,3. Esta posibilidad pronto atrajo el interés en los aspectos éticos, con base en la controversia derivada del beneficio de conocer la susceptibilidad a un padecimiento para el cual aún no existe tratamiento curativo y no es posible modificar el desenlace clínico4,5. Específicamente en el caso de la SCA, en el año 2010 se emitieron lineamientos por parte de la European Molecular Quality Genetics Network (EMQN) que describen los requerimientos para la realización de análisis presintomático en laboratorios afiliados a esta, con la finalidad de garantía de calidad6. Sin embargo, no existe un estándar que oriente sobre la realización de DPS en sujetos con riesgo de presentar esta enfermedad y en la actualidad se fundamenta en lineamientos éticos generales y otros diseñados para padecimientos como HD.
ObjetivoAnalizar las consideraciones éticas, procedimientos de asesoramiento genético y recomendaciones derivadas de los estudios que realizaron DPS de sujetos con riesgo de SCA.
DesarrolloEl DPS se define como el estudio que identifica a sujetos sanos que desarrollarán un trastorno genético si llegan a vivir lo suficiente7. Se considera que el DPS comenzó en 1983 con la identificación de sujetos en riesgo de desarrollar HD8. Debido a su origen y modo de herencia, la SCA se unió a este grupo de enfermedades neurodegenerativas que pueden ser diagnosticadas aun antes de la presentación de síntomas9.
Ataxias espinocerebelosasLas SCA son un grupo de trastornos neurodegenerativos con patrón de herencia autosómico dominante, con síntomas causados por la disfunción del cerebelo y el tronco cerebral, así con sus vías y conexiones asociadas10,11. La incidencia de la SCA se estima en 2-3 casos por 100.000 habitantes12. Ruano et al. mediante un metaanálisis determinaron la prevalencia de SCA autosómica dominante entre 0 y 5,6 por 100.000 habitantes13, de igual forma Polo et al. en Cantabria identificaron una prevalencia de 0,29 casos por 100.000 habitantes14. Desde su identificación se han utilizado distintas clasificaciones, siendo la basada en los loci génicos la más aceptada15. Hasta la fecha más de 35 tipos han sido descritos16, y destacan las originadas por los repetidos CAG: SCA1, SCA2, SCA3, SCA6, SCA7, SCA12, SCA17 y DPRLA; las cuales son responsables de más del 50% de los casos. Otras SCA tienen su origen en distintos tipos de repetidos, mutaciones puntuales y deleciones17,18.
Las SCA autosómicas dominantes se caracterizan clínicamente por la presencia de ataxia cerebelosa progresiva, que puede asociar oftalmoplejía, signos piramidales, extrapiramidales, sensitivos, deterioro cognitivo y neuropatía periférica. Habitualmente los síntomas se manifiestan en la edad adulta, sin embargo, varios tipos presentan fenómeno de anticipación. Debido a la heterogeneidad clínica y genética de este grupo de enfermedades es necesaria su identificación mediante estudio molecular en ADN genómico obtenido de una muestra de sangre15. No obstante, aún no se ha desarrollado un tratamiento curativo para este grupo de enfermedades. El manejo actual se enfoca al tratamiento sintomático y de las complicaciones, además de brindar un asesoramiento genético adecuado por parte de un equipo multidisciplinario19,20.
MetodologíaCon la finalidad de analizar las consideraciones éticas y procedimientos de asesoramiento genético en el DPS de sujetos en riesgo de presentar SCA, se realizó una búsqueda en Pubmed de las publicaciones con la combinación de palabras clave «asesoramiento genético en SCA» y «diagnóstico presintomático y SCA». La búsqueda mostró 12 artículos, de los cuales fueron eliminados aquellos que solo describieron el DPS como parte de un estudio familiar, aquellas que no realizaron procedimientos de asesoramiento genético antes del análisis y también aquellos en las que no existía una identificación clara en el número de sujetos con riesgo de SCA de otros trastornos neurodegenerativos. La información es limitada y solamente 6 trabajos cumplieron con estos requisitos. Adicionalmente se realizó una búsqueda de cada una de las publicaciones que sirvieron como base para las consideraciones éticas, con la finalidad de conocer el origen de estas.
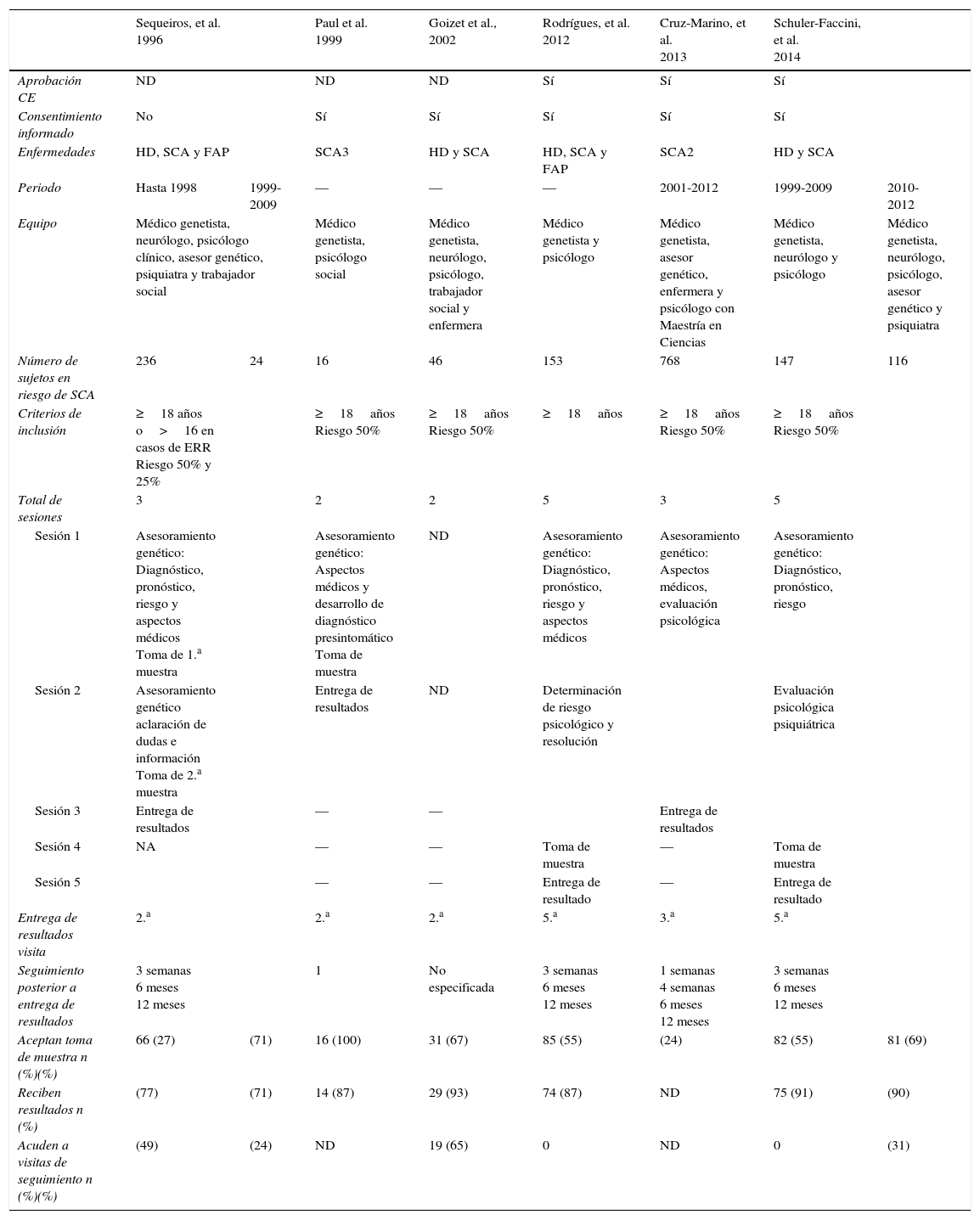
EstudiosCada una de las publicaciones analizadas en la presente revisión tomó en cuenta lineamientos disponibles en su momento para la realización del DPS dentro de la investigación. Debido a la similitud en la presentación clínica y evolución de otros trastornos neurodegenerativos, algunos de los trabajos realizan estudios en más de un trastorno. Solo Paul et al. y Cruz-Marino et al. consideraron la inclusión exclusiva de sujetos en riesgo de SCA3 y SCA2 respectivamente21,22. Goizet et al. y Schuler-Faccini et al. estudiaron sujetos con riesgo de HD y SCA23,24; Sequeiros et al. (1996 a,b)24, y Rodrigues et al. llevaron a cabo sus estudios en sujetos con riesgo de HD, SCA y polineuropatía familiar amiloide8. Aun con la diferencia entre las enfermedades estudiadas en cada uno de los trabajos, existió similitud en los procedimientos realizados en el DPS lo cual hace posible su comparación.
Uno de los primeros aspectos a considerar cuando se realiza un DPS es el derecho de cualquier sujeto (asintomático) a no conocer su condición, en especial si no existe beneficio médico, ya que esto causa un cambio en la percepción de sí mismo y podría ser perjudicial para el sujeto, con la posibilidad de desencadenar efectos catastróficos como se ha registrado en enfermedades como HD25. Por este motivo las buenas prácticas clínicas en investigación y los lineamientos emitidos por distintos organismos requieren la realización de un adecuado proceso de consentimiento informado previo al DPS. Éste debe puntualizar aspectos importantes como la confidencialidad, riesgos, beneficios, limitaciones del análisis, entre otros. La mayoría de los estudios analizados consideró la firma de un consentimiento informado como parte del proceso de asesoramiento genético. Es importante que con la finalidad del resguardo de la confidencialidad se analice en dónde permanecerán los resultados y si estos deben ser archivados por separado, permaneciendo solo disponibles para el interesado. Otro aspecto a considerar en este apartado es la madurez del sujeto que recibirá la información. Sin embargo, para determinar esta capacidad sería necesario analizar cada caso en particular. A nivel global se pensaría que un sujeto cumpla con la mayoría de edad está preparado para la toma de decisiones en este ámbito, sin embargo, el estado de mayoría legal varía dependiendo de cada país con extremos desde 14 hasta los 21 años. Debido a esto, por consenso, se ha determinado que la edad mínima para decidir la participación en DPS debe ser de 18 años5. Con respecto a la edad, solamente el estudio de Sequeiros et al. (1996)24 brindó la oportunidad de analizar a sujetos de 16 años o mayores como parte de un protocolo de estimación de riesgo reproductivo. La edad mínima para la realización del DPS puede ser un punto de controversia y sería deseable analizar el estado de madurez del individuo previo al DPS para asegurar su bienestar. Por otro lado debe respetarse el derecho de cada sujeto a decidir libremente si desea conocer o no su condición de salud; por lo que el proceso de DPS debería proporcionar los elementos para que el individuo tome la decisión de forma independiente y considerando solo como un referente la edad de 18 años.
Esquemas de asesoramiento genéticoDistintos esquemas han sido implementados, todos ellos con apoyo de equipos multidisciplinarios. Es necesario brindar a los sujetos la oportunidad de recibir la mejor calidad de asesoramiento genético y soporte, tanto médico como psicológico, para afrontar el DPS. De esta forma los estudios revisados contaron por lo menos con un médico genetista y un psicólogo, quienes tenían la responsabilidad básica de brindar información sobre el diagnóstico, riesgos, pronóstico, aspectos médicos, así como la responsabilidad de evaluar la integridad psicológica. Otros incluían también asesor genético, psiquiatra, neurólogo, enfermera y trabajador social. La información detallada se resume en la tabla 1. En el caso del trabajo de Schuler-Faccini et al. fue posible el análisis de un mismo esquema de asesoramiento con variantes en el equipo multidisciplinario, ya que en su etapa inicial contaban solamente con un médico genetista, un psicólogo y un neurólogo; más tarde integraron también un asesor genético y un psiquiatra. De esta forma es posible hacer la comparación entre los periodos de 1999-2009 y 2010-201224, siendo evidente un ligero incremento en el porcentaje de los sujetos que aceptaron realizar el DPS, así como crecimiento importante en el porcentaje de estos que cumplieron con las visitas de seguimiento. Con respecto a la formación académica de los miembros que deben integrar el equipo multidisciplinario, no existe un mínimo de especialidades. Es deseable que para brindar la mejor atención estén constituidos por al menos un médico genetista y un neurólogo, que tendrían responsabilidad sobre aspectos médicos como el diagnóstico, análisis clínico, manejo y tratamiento de la enfermedad y sus complicaciones. Por otra parte, es también indispensable el apoyo de un psicólogo y/o psiquiatra y un asesor genético quienes tendrán la responsabilidad en la determinación de la madurez, integridad psicológica, riesgos derivados del DPS, así como en el soporte para afrontar el diagnóstico y acompañamiento en el proceso. No obstante, para brindar los servicios óptimos en el asesoramiento genético pueden ser necesarios profesionales de otras especialidades como enfermeras, trabajadoras sociales, nutricionistas, entre otras7.
Comparación de las características de los estudios que realizaron diagnóstico presintomático en sujetos con riesgo de SCA
| Sequeiros, et al. 1996 | Paul et al. 1999 | Goizet et al., 2002 | Rodrígues, et al. 2012 | Cruz-Marino, et al. 2013 | Schuler-Faccini, et al. 2014 | |||
|---|---|---|---|---|---|---|---|---|
| Aprobación CE | ND | ND | ND | Sí | Sí | Sí | ||
| Consentimiento informado | No | Sí | Sí | Sí | Sí | Sí | ||
| Enfermedades | HD, SCA y FAP | SCA3 | HD y SCA | HD, SCA y FAP | SCA2 | HD y SCA | ||
| Periodo | Hasta 1998 | 1999-2009 | — | — | — | 2001-2012 | 1999-2009 | 2010-2012 |
| Equipo | Médico genetista, neurólogo, psicólogo clínico, asesor genético, psiquiatra y trabajador social | Médico genetista, psicólogo social | Médico genetista, neurólogo, psicólogo, trabajador social y enfermera | Médico genetista y psicólogo | Médico genetista, asesor genético, enfermera y psicólogo con Maestría en Ciencias | Médico genetista, neurólogo y psicólogo | Médico genetista, neurólogo, psicólogo, asesor genético y psiquiatra | |
| Número de sujetos en riesgo de SCA | 236 | 24 | 16 | 46 | 153 | 768 | 147 | 116 |
| Criterios de inclusión | ≥18 años o>16 en casos de ERR Riesgo 50% y 25% | ≥18años Riesgo 50% | ≥18años Riesgo 50% | ≥18años | ≥18años Riesgo 50% | ≥18años Riesgo 50% | ||
| Total de sesiones | 3 | 2 | 2 | 5 | 3 | 5 | ||
| Sesión 1 | Asesoramiento genético: Diagnóstico, pronóstico, riesgo y aspectos médicos Toma de 1.a muestra | Asesoramiento genético: Aspectos médicos y desarrollo de diagnóstico presintomático Toma de muestra | ND | Asesoramiento genético: Diagnóstico, pronóstico, riesgo y aspectos médicos | Asesoramiento genético: Aspectos médicos, evaluación psicológica | Asesoramiento genético: Diagnóstico, pronóstico, riesgo | ||
| Sesión 2 | Asesoramiento genético aclaración de dudas e información Toma de 2.a muestra | Entrega de resultados | ND | Determinación de riesgo psicológico y resolución | Evaluación psicológica psiquiátrica | |||
| Sesión 3 | Entrega de resultados | — | — | Entrega de resultados | ||||
| Sesión 4 | NA | — | — | Toma de muestra | — | Toma de muestra | ||
| Sesión 5 | — | — | Entrega de resultado | — | Entrega de resultado | |||
| Entrega de resultados visita | 2.a | 2.a | 2.a | 5.a | 3.a | 5.a | ||
| Seguimiento posterior a entrega de resultados | 3 semanas 6 meses 12 meses | 1 | No especificada | 3 semanas 6 meses 12 meses | 1 semanas 4 semanas 6 meses 12 meses | 3 semanas 6 meses 12 meses | ||
| Aceptan toma de muestra n (%)(%) | 66 (27) | (71) | 16 (100) | 31 (67) | 85 (55) | (24) | 82 (55) | 81 (69) |
| Reciben resultados n (%) | (77) | (71) | 14 (87) | 29 (93) | 74 (87) | ND | 75 (91) | (90) |
| Acuden a visitas de seguimiento n (%)(%) | (49) | (24) | ND | 19 (65) | 0 | ND | 0 | (31) |
CE: comité de ética; ERR: estimación de riesgo reproductivo; FPA: polineuropatía familiar amiloide; HD: enfermedad de Huntington; NA: no aplica; ND: no disponible; SCA: ataxia espinocerebelosa.
De acuerdo con los lineamientos disponibles se recomienda llevar a cabo al menos 3 sesiones de asesoramiento previas al DPS para la protección de riesgos de los individuos7. Con esta base cada uno de los estudios programó un número variable de visitas previas al DPS. El mínimo de sesiones programadas fue de 2. Estas otorgaron el asesoramiento en el primer contacto, obteniendo la muestra en la misma visita; en una visita posterior se realizó la entrega de los resultados, como sucedió en el estudio de Goizet et al.. A diferencia de estos, otros estudios como los de Rodrigues et al. y Schuler-Faccini et al. optaron por esquemas conservadores en los cuales se brindó una atención más detallada, con énfasis en el análisis de la integridad y riesgo psicológico, dando oportunidad a la resolución de trastornos detectados y, posteriormente, a realizar la toma de muestra para DPS, concluyendo con la entrega del resultado en la visita final. Werts et al. recomiendan 3 sesiones previas a la entrega de los resultados, sin embargo, no debe limitarse solo a estas y deberá evaluarse en equipo cada caso con especial atención en la integridad psicológica con la finalidad de minimizar los riesgos de eventos catastróficos.
De la misma forma se recomiendan las sesiones de seguimiento posteriores a la entrega de los resultados del DPS en los sujetos. En el caso de sujetos con resultado desfavorable se sugieren sesiones sin un límite determinado y para los sujetos con resultado favorable también con la finalidad de eliminar los sentimientos de culpa7. De acuerdo a este punto, los trabajos de investigación revisados contaron con programas de seguimiento de por lo menos un contacto posterior a la entrega de resultados a excepción del trabajo de Goizet et al. en el que no se hace referencia. La mayoría de los autores diseñaron sus trabajos de acuerdo a un programa de seguimientos múltiples de 3-4 visitas (tabla 1), además de ofrecer visitas posteriores a requerimiento de los sujetos. Sin embargo, el porcentaje de sujetos referido por las publicaciones que acudieron a los seguimientos programados fue del 0-49% en el mejor de los casos. Todos los reportes coinciden en la baja tasa de recurrencia a las visitas de seguimiento, sin embargo, no se identifica una causa, salvo en el estudio de Cruz-Marino et al., por lo que es necesario que durante el DPS se enfatice al paciente sobre la importancia del seguimiento, ya que este le brindará la oportunidad de estar en contacto para prevenir complicaciones y conocer la información pertinente en caso de existir nuevas opciones de tratamiento.
Lineamientos para diagnósticos presintomáticosLos lineamientos emitidos por la Organización Mundial de la Salud en 2003, recomiendan analizar algunos puntos básicos para la consideración del DPS, como: (i) Garantizar la confidencialidad, (ii) Brindar información con relación a las limitaciones del análisis, (iii) No presentar enfermedades mentales al momento de realizar el análisis, (iv) Evidencia de que los resultados del análisis serán de utilidad para disminuir daño, y (v) Compromiso de que el análisis estará acompañado de un programa con asesoramiento adecuado. Adicionalmente debe de existir un consentimiento informado7.
En el caso de la SCA se han emitido requisitos por parte de organismos como la EMQN) para la realización de DPS en los laboratorios afiliados. Entre estos destacan los siguientes: (i) No se realiza DPS en menores de edad si no existe un beneficio, (ii) Es esencial la documentación del proceso de asesoramiento genético y del seguimiento, (iii) Los requerimientos de DPS deberán ser solicitados por el grupo que brindó el asesoramiento, además de estar acompañados de los documentos que avalen que el asesoramiento se realizó, (iv) Consentimiento informado de acuerdo a normas locales y/o internacionales, y (v) Deberá existir información de presentación familiar, entre otras6.
A diferencia de lo que sucede en las SCA, para la HD después de varias reuniones en 1994, en una labor conjunta, la Asociación Internacional de Enfermedad de Huntington (IHA) y la Federación Mundial de Neurología (WFN) establecieron los lineamientos de diagnóstico predictivo para esta enfermedad26. Las recomendaciones no solo han mostrado con éxito ser el parámetro mínimo a considerar en los pacientes con HD sino que han revelado ser útiles también en otras enfermedades neurodegenerativas como la demencia frontotemporal y la SCA5. El análisis de la información generada en las publicaciones revisadas, en conjunto con los lineamientos existentes para otras enfermedades que han mostrado ser de utilidad, podrían ser la base para generar recomendaciones específicas en el DPS de SCA, entre las que se incluya el registro de los resultados de estas acciones, para la evaluación del beneficio de su aplicación.
ConclusionesEl DPS en SCA brinda a los sujetos con riesgo la posibilidad de saber si presentarán el padecimiento en la edad adulta, además de permitirles la planificación familiar con base en el conocimiento de su condición de riesgo. El DPS debe ser realizado bajo lineamientos que salvaguarden el bienestar físico y psicológico de los sujetos. Hasta la fecha de esta revisión no se cuenta con lineamientos específicos para la realización de DPS en pacientes con riesgo de padecer SCA, fundamentando el DPS en lineamientos emitidos por organismos internacionales y otros específicamente diseñados para otras enfermedades. Es necesaria la implementación de esquemas de asesoramiento genético en todos aquellos centros que pretenden realizar diagnóstico de SCA antes de la presentación de síntomas. Es fundamental que un comité de ética apruebe, regule y garantice el cumplimiento de los procedimientos en caso del DPS, así como la participación de los ministerios de salud con apoyo de organizaciones especializadas, en caso de estar disponibles. Dentro de lo posible, el análisis molecular para diagnóstico de SCA debería realizarse solamente a pacientes con clínica sugestiva, mayores de 18 años y con un riesgo mínimo del 50%, es decir que un progenitor esté afectado, además de contar con programas de asesoramiento genético integrado por un grupo multidisciplinario, el cual debería incluir al menos un asesor genético, un médico genetista, un neurólogo y un psicólogo y/o psiquiatra.
FinanciaciónEl presente trabajo no ha contado con financiamiento de ningún organismo público o privado.
Conflicto de interesesNo existe conflicto de intereses de ninguno de los autores.
Dra. en C. María Cristina Moran Moguel
El presente trabajo no ha sido presentado en la reunión anual de SEN ni en otro foro.

