




Desde mitad del siglo xx se ha apuntado a la importancia de la amígdala en la epilepsia, aunque los mecanismos básicos de esta participación en su mayoría son aún desconocidos. Esta ignorancia es aún mayor cuando se tienen en cuenta las distintas subdivisiones de la amígdala, especialmente sus partes mediales. En este trabajo evaluamos la participación de la amígdala extendida medial en un modelo animal de epilepsia, así como las consecuencias que tiene el epileptógeno en esta estructura cerebral.
Material y métodosSe utilizaron ratas adultas Wistar machos (n=48); 24 animales recibieron inyecciones intraperitoneales de pentilentetrazol y 24, de salina. Luego de 2, 6, 12 y 24 h de sobrevida, los animales se fijaron, y sus cerebros se cortaron seriadamente y se procesaron para fos (inmunoquímica) y muerte neuronal con la técnica A-Cu-Ag. Los datos se analizaron con un ANOVA de 2 vías seguido de un test post-hoc (LSD de Fisher).
ResultadosMuy poca activación fos se halla en animales controles. En animales experimentales, fos fue rápidamente inducida en la amígdala extendida medial a las 2 h. Esta activación fue sostenida hasta las 12 h y retornó a valores basales a las 24 h. Sin embargo, el estado epiléptico no produjo muerte neuronal.
ConclusionesSe demuestra así una participación de la amígdala extendida medial en mecanismos epilépticos en los cuales subyace un componente inhibitorio. Sin embargo, el estado epiléptico inducido no produce muerte neuronal en esta estructura.
Since middle of the 20th century the importance of amygdala in epilepsy it has suggested, although the basic mechanisms of this participation are still unknown. This ignorance increases when the different subdivisions of amygdala are considered, especially the medial amygdala. In this work we assess the involvement of the medial extended amygdala in an animal model of epilepsy and the consequences of its application in this brain structure.
Material and methodsForty eight adult Wistar male rats were used, of which 24 of them received i.p. injections of pentylenetetrazole, and 24 (controls) were injected with saline. After 2, 6, 12 and 24 h survival, animals were fixed; the brains were sectioned serially and stained for fos (immunochemistry) and for neuronal death with the A-Cu-Ag technique. Data were analysed using two-way ANOVA followed by the Fisher post hoc test.
ResultsVery few or no fos-immunoreactive neurons were seen in control animals. In experimental animals, fos was rapidly induced in structures of medial extended amygdala with peak levels at 2 h. Marked fos immunoreactivity persisted up to 12 h followed by a gradual return to baseline at 24 h. However, status epilepticus did not induced neuronal death.
ConclusionsThese results show involvement of medial extended amygdala in epileptic mechanisms with an inhibitory component. However, neuronal death is not a consequence of status epilepticus-induced by pentylentetrazole.
El conocimiento de los mecanismos celulares y moleculares subyacentes a los distintos tipos de epilepsias es todavía limitado. Actualmente, se desconocen los factores que transforman una región del cerebro normal en epiléptico, por lo que el estudio de animales que presentan epilepsia resulta muy útil para el conocimiento de la fisiopatología de este grupo de enfermedades1.
El modelo animal más utilizado para estudiar la epilepsia es el modelo de estado epiléptico (EE). Para provocar el EE, se utilizan diferentes agentes convulsivos como el ácido kaínico2, la pilocarpina3 y el pentilentetrazol (PTZ)4 para inducir una fase aguda de EE caracterizado por crisis tonicoclónicas que no ceden. El problema básico en epileptología experimental consiste en determinar las condiciones que, en cada caso, alteran el equilibrio entre excitación e inhibición neuronal.
Por otra parte, la estructura anatómica denominada amígdala extendida medial (AMeXM) desarrollada por nuestro laboratorio5 es un área del cerebro que no se ha estudiado en profundidad en el ámbito de la epilepsia experimental. Ésta consiste en dos subdivisiones mayores, una, la amígdala central y otra que incluye la amígdala medial y el núcleo intersticial de la estría terminal medial (NIETm), denominada división medial. El sistema de la AMeXM participa en el procesamiento de señales feromonales con efectos en el sistema endocrino y los aspectos emocionales y reproductivos de la conducta6,7.
Numerosos informes involucran al complejo amigdalino en enfermedades como la epilepsia8, la enfermedad de Alzheimer9, la depresión10 y la ansiedad11. Específicamente, en el campo de la epilepsia, el interés en la amígdala como una estructura asociada a la epilepsia surgió en la década de los cincuenta, cuando se halló un considerable daño neuronal en pacientes que habían sufrido un EE. Los modelos animales han puesto de manifiesto que la amígdala es susceptible de ser dañada en el estado epiléptico y que éste produce cambios histopatológicos12,13. Sin embargo, muy poco se conoce acerca de los detalles de la distribución y los cambios temporales de estos cambios en la AMeXM.
Un modo de evaluar estos cambios es mediante el estudio de la expresión de genes tempranos14. Desde su descubrimiento como un gen de transcripción temprana, c-fos se ha utilizado como una herramienta de cartografía funcional y anatómica debido a que identifica células y circuitos neuronales que se activan en respuesta a diferentes estímulos14. En este trabajo, se ha procurado revelar el patrón de activación con la expresión del gen c-fos, la proteína fos, luego de la inducción del EE con PTZ. Asimismo, se realizó un estudio para revelar posible muerte neuronal como efecto de la aplicación del epileptógeno. El PTZ se ha utilizado frecuentemente en animales como un modelo de epilepsia generalizada; uniéndose al sitio para picrotoxina en el receptor GABAA, produce daño en la actividad del canal de cloro y así bloquea la actividad inhibitoria mediada por el GABA15.
Material y métodosÁmbito de estudioEste trabajo fue llevado a cabo en el Laboratorio de Neurobiología del Sistema Límbico del Instituto de Investigaciones Médicas Mercedes y Martín Ferreyra.
Período de estudioEn febrero de 2006 se inició con los trabajos experimentales y finalizó, al cabo de 12 meses, en marzo de 2007.
AnimalesSe utilizaron 48 ratas Wistar machos adultos (200-250g), aislados en habitación ad-hoc 48h antes del experimento en condiciones generales de bioterio. Todos los procedimientos fueron realizados de acuerdo con el Real Decreto 1201/2005 sobre la protección de los animales utilizados.
Inducción del estado epiléptico con PTZSe inyectó a los animales (n = 6) con PTZ intraperitoneal diluido en solución salina al 0,9% de acuerdo al esquema de Pineau et al16. Sólo se consideró en este estudio a los animales que llegaron al estado 4. Paralelamente, animales controles recibieron de igual manera solución salina.
Fijación del cerebroA las 2, 6, 12 y 24h tras la inducción del EE, se anestesió a los animales con hidrato de cloral al 30%, para luego perfundir 100ml de solución lavadora y finalmente 300ml de paraformaldehído al 4%. Luego el cerebro se colocó en solución de sucrosa al 30%; 48h después se cortó a 40μm en un microtomo de congelación y se recolectaron seriadamente en la misma solución fijadora.
Técnica inmunoquímicaPara la detección de la proteína fos las secciones de cerebros se lavaron en buffer (PO4, 0,01mol) y se inhibió la peroxidasa interna. Los cortes se lavaron y se procedió al bloqueo de uniones inespecíficas, que se incubaron en suero normal de caballo al 5%. Luego, los cortes se incubaron en anticuerpo primario (fos: 1/5.000, sc-52, Santa Cruz), para luego incubarse por 2h en anticuerpo secundario biotinilado (1/200, Vector), 2h con avidina-biotina-peroxidasa (Vector) y la reacción se reveló con diaminobenzidina como cromógeno agregando cloruro de cobalto que confiere a la reacción color negro. Los cortes fueron montados y cubiertos con DPX.
Técnica amino-cupro-argénticaEl método de impregnación A-Cu-Ag17 se seleccionó debido a que se aplica habitualmente en el laboratorio y es la última y mejorada versión de los métodos cupro-argénticos.
Análisis cuantitativo celularLos cortes se analizaron con microscopio Axioplan con cámara Leica adaptada. El análisis cuantitativo celular se realizó con el programa Scion Image (Scion Corporation, 2000, NIH, Estados Unidos) en cortes coronales a un aumento de ×20.
Se seleccionaron para el análisis las siguientes estructuras de la AMeXM18: núcleo amigdalino medial (Me) en los planos con respecto al bregma: –1,80 y –2,56 para el Me anterodorsal (MeAD), –2,30 para el Me anteroventral (MeAV), –3,14 y –3,60 para el Me posterodorsal (MePD) y –3,14 para el Me posteroventral (MePV); el NIETm en dos planos con respecto al bregma: –0,30 para el NIET anterior (NIETa) y ventral (NIETv) y –0,80 para el NIET posterior (NIETp).
Análisis estadísticoLos datos fueron analizados usando un análisis de la variancia (ANOVA) de dos factores, seguido por un test post-hoc LSD de Fisher; los valores se expresaron como las medias de cada grupo ± el error estándar, y se consideró una p < 0,05 como límite de significación estadística.
ResultadosExpresión de fos tras de la inyección de PTZEn todos los animales controles inyectados con solución salina se aprecian pocos y dispersos núcleos de células activados en las áreas aquí evaluadas.
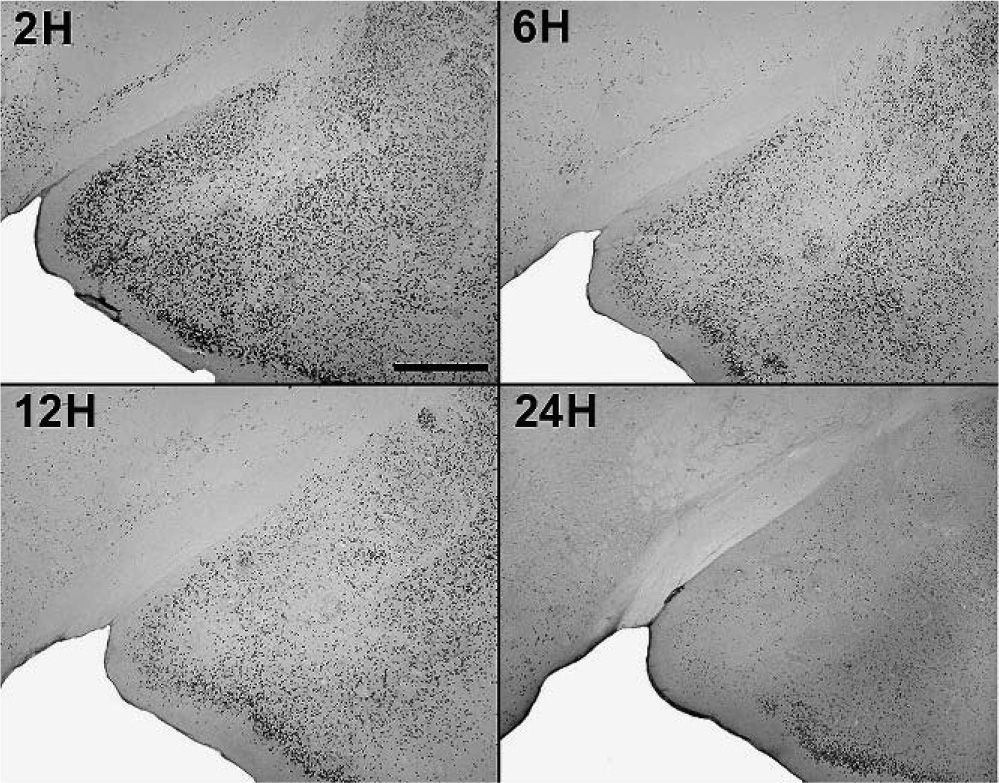
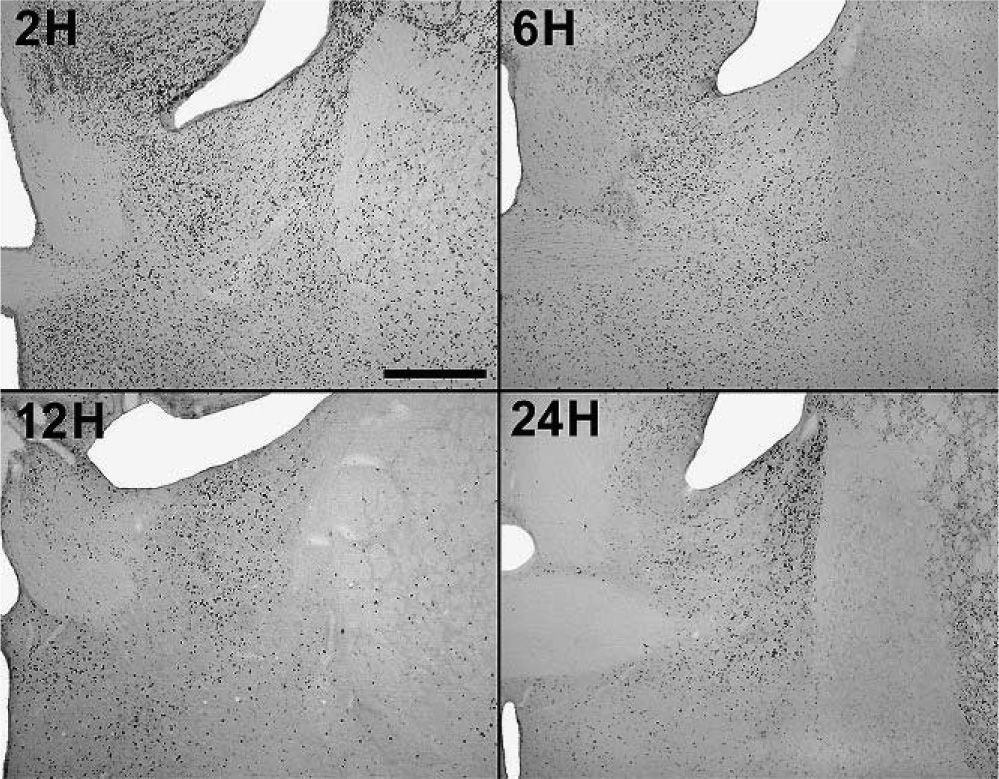
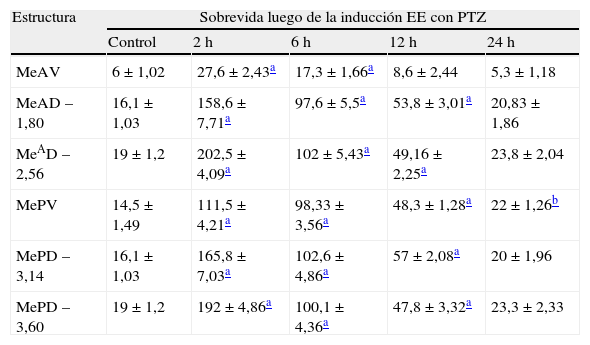
En animales experimentales, la mayor expresión de la proteína fos se encuentra en aquellos cuya sobrevida es de 2h tras la inyección, donde las diferencias con los animales controles son estadísticamente significativas para todas las áreas donde se llevó c cabo el análisis cuantitativo celular de núcleos fos (+) (tablas 1 y 2). El MeAD (tabla 1) y el NIETp (tabla 2) son las áreas donde se puede observar una mayor activación en comparación con los controles. El MePD (fig. 1) y el NIETa (fig. 2) se encuentran, asimismo, profusamente marcados para la proteína fos. En todas las áreas bajo evaluación, se aprecian numerosos núcleos celulares que se han teñido con el anticuerpo para fos. Se destacan, entre otros, la diferenciación de las tres columnas en el NIETp (5), una considerable cantidad de células activadas en todas las divisiones del Me en la región cercana al tracto olfatorio accesorio y el tracto óptico (fig. 1) y el NIETv (fig. 2) bajo la comisura anterior.
Análisis cuantitativo de núcleos inmunorreactivos para fos en las subdivisiones del núcleo amigdalino medial después de la inducción del estado epiléptico (EE) con pentilentetrazol (PTZ)
| Estructura | Sobrevida luego de la inducción EE con PTZ | ||||
| Control | 2h | 6h | 12h | 24h | |
| MeAV | 6 ± 1,02 | 27,6 ± 2,43a | 17,3 ± 1,66a | 8,6 ± 2,44 | 5,3 ± 1,18 |
| MeAD –1,80 | 16,1 ± 1,03 | 158,6 ± 7,71a | 97,6 ± 5,5a | 53,8 ± 3,01a | 20,83 ± 1,86 |
| MeAD –2,56 | 19 ± 1,2 | 202,5 ± 4,09a | 102 ± 5,43a | 49,16 ± 2,25a | 23,8 ± 2,04 |
| MePV | 14,5 ± 1,49 | 111,5 ± 4,21a | 98,33 ± 3,56a | 48,3 ± 1,28a | 22 ± 1,26b |
| MePD –3,14 | 16,1 ± 1,03 | 165,8 ± 7,03a | 102,6 ± 4,86a | 57 ± 2,08a | 20 ± 1,96 |
| MePD –3,60 | 19 ± 1,2 | 192 ± 4,86a | 100,1 ± 4,36a | 47,8 ± 3,32a | 23,3 ± 2,33 |
MeAD: núcleo amigdalino medial anterodorsal; MeAV: núcleo amigdalino medial anteroventral; MePD: núcleo amigdalino medial posterodorsal; MePV: núcleo amigdalino medial posteroventral.
Se utilizó un ANOVA de dos factores seguido por un test post-hoc LSD de Fisher. Los datos son expresados como la media ± el error estándar de la media.
Análisis cuantitativo de núcleos inmunorreactivos para fos en las subdivisiones del NIET medial después de la inducción del estado epiléptico (EE) con pentilentetrazol (PTZ)
| Estructura | Sobrevida luego de la inducción EE con PTZ | ||||
| Control | 2h | 6h | 12h | 24h | |
| NIETa | 13,6 ± 1,59 | 167,6 ± 6,4* | 126,5 ± 9,68* | 50,1 ± 5,32* | 16,5 ± 1,46 |
| NIETp | 18,3 ± 1,08 | 207,5 ± 8,42* | 132,6 ± 5,68* | 55,5 ± 3,97* | 21,5 ± 2,87 |
| NIETv | 12,1 ± 1,48 | 84,16 ± 3,51* | 62,5 ± 2,26* | 45 ± 2,5* | 11,16 ± 0,91 |
NIET: núcleo intersticial de la estría terminal; NIETa: núcleo intersticial de la estría terminal anterior; NIETp: núcleo intersticial de la estría terminal posterior; NIETv: núcleo intersticial de la estría terminal ventral.
Se utilizó un ANOVA de dos factores seguido por un test post-hoc LSD de Fisher. Los datos son expresados como la media ± el error estándar de la media.
 que muestran la activación en el núcleo amigdalino medial posterodorsal y el posteroventral a distintas sobrevidas luego de la inyección de pentilentetrazol. Escala barra: 500μm.")
 que muestran la activación en el núcleo intersticial de la estría terminal anterior y ventral a distintas sobrevidas luego de la inyección de pentilentetrazol. Escala barra: 500μm.")
A partir de una sobrevida de 6h, si bien la marcación disminuye, las diferencias continúan siendo significativas al igual que en los animales sacrificados 12h luego de la inyección de PTZ. Nuevamente las áreas anteriormente mencionadas son las que presentan mayor activación (figs. 1 y 2; tablas 1 y 2). Por último, en animales cuya sobrevida llegó a las 24h se puede observar que las diferencias entre controles y experimentales no son significativas, ya que la activación llega a valores basales comparables con los de los controles (tablas 1 y 2).
El ANOVA llevado a cabo pone de manifiesto lo dicho anteriormente, esto es, que el pico de activación se encuentra a las 2h y que luego la marcación disminuye paulatinamente en animales con mayor tiempo de sobrevida (figs. 3 y 4). De destacar es el hecho de que las diferencias entre los distintos grupos experimentales son significativas para la mayor parte de las estructuras en el análisis cuantitativo celular (tablas 1 y 2).
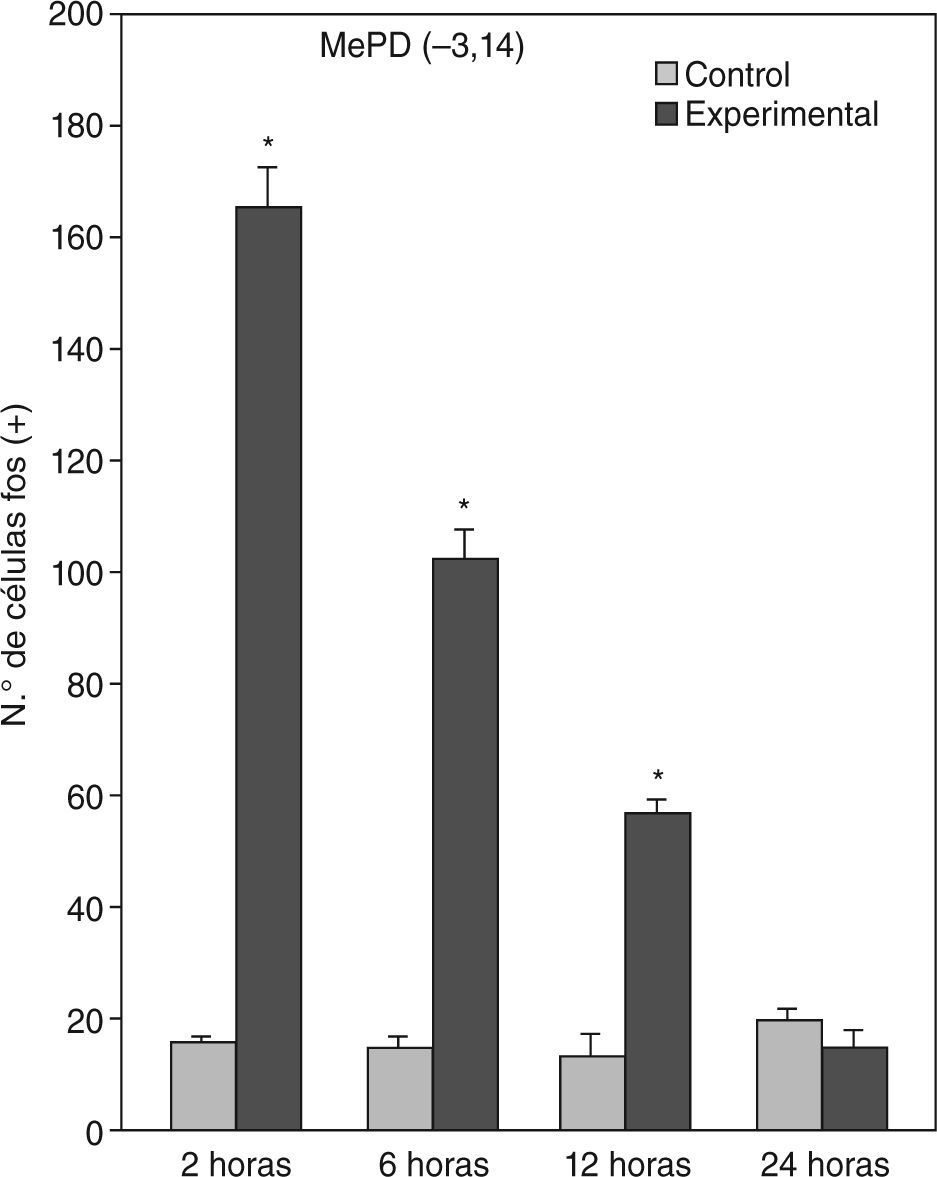
, cuyos factores son el “tratamiento” y las diferentes “sobrevidas”. El resultado del estudio de los factores es para tratamiento: F(1,40) = 1.111; p < 0,001; para sobrevida: F(3,40) = 220,3; p < 0,001; interacción: F(3,40) = 209,4; p < 0,001. Los resultados son expresados como la media ± el error estándar (*p < 0,001).")
Análisis cuantitativo celular de núcleos fos+ de animales con sobrevidas de 2, 6, 12 y 24h luego de la aplicación de pentilentetrazol y posterior ANOVA de dos vías en el núcleo amigdalino medial posterodorsal (MePD), cuyos factores son el “tratamiento” y las diferentes “sobrevidas”. El resultado del estudio de los factores es para tratamiento: F(1,40) = 1.111; p < 0,001; para sobrevida: F(3,40) = 220,3; p < 0,001; interacción: F(3,40) = 209,4; p < 0,001. Los resultados son expresados como la media ± el error estándar (*p < 0,001).
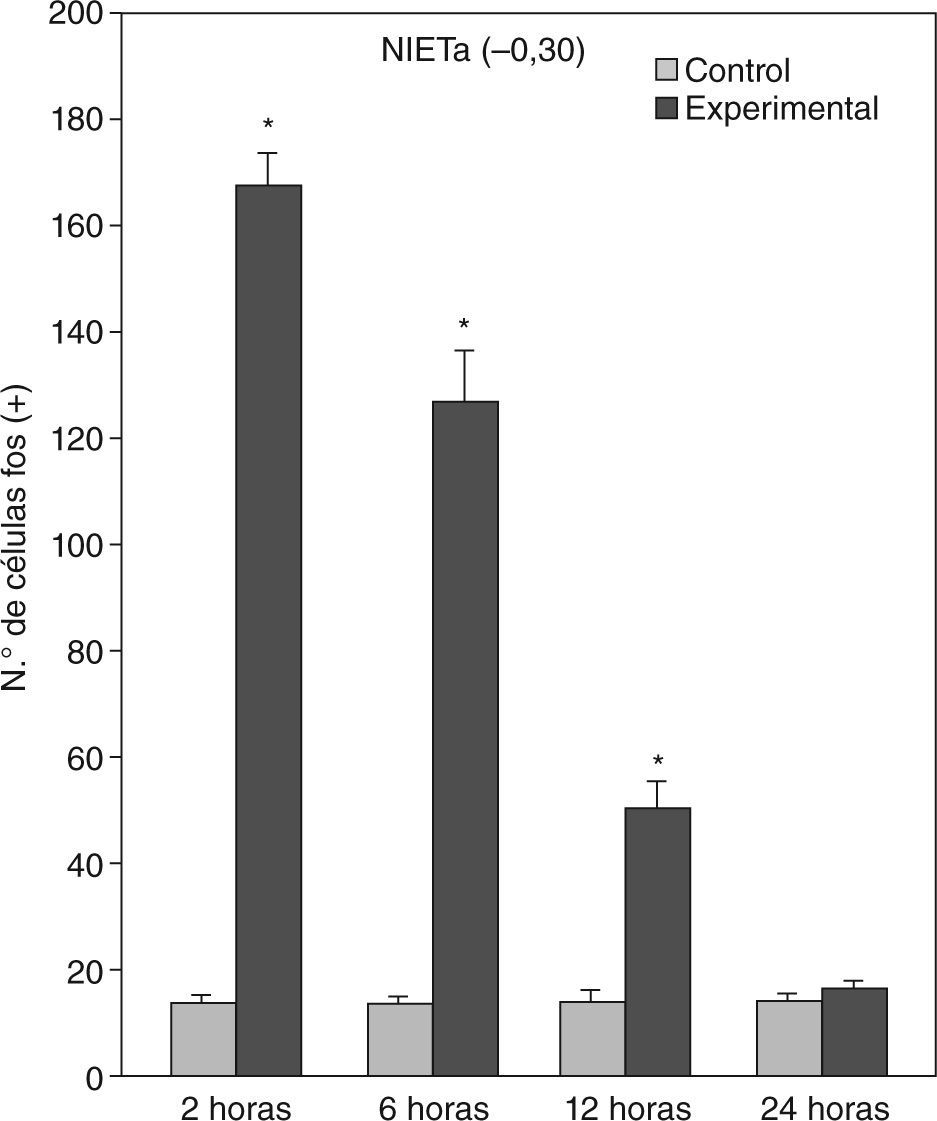
 cuyos factores son el “tratamiento” y las diferentes “sobrevidas”. El resultado del estudio de los factores es para tratamiento: F(1,40) = 640,46; p < 0,001; para sobrevida: F(3,40) = 130,34; p < 0,001; interacción: F(3,40) = 132,90; p < 0,001. Los resultados son expresados como la media ± el error estándar (*p < 0,001).")
Análisis cuantitativo celular de núcleos fos+ de animales con sobrevidas de 2, 6, 12 y 24h luego de la aplicación de pentilentetrazol y posterior ANOVA de dos vías en el núcleo intersticial de la estría terminal anterior (NIETa) cuyos factores son el “tratamiento” y las diferentes “sobrevidas”. El resultado del estudio de los factores es para tratamiento: F(1,40) = 640,46; p < 0,001; para sobrevida: F(3,40) = 130,34; p < 0,001; interacción: F(3,40) = 132,90; p < 0,001. Los resultados son expresados como la media ± el error estándar (*p < 0,001).
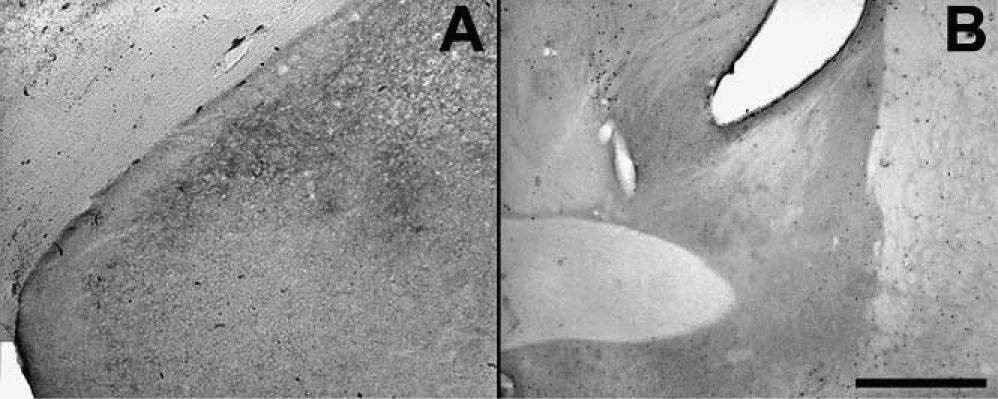
Bajo el paradigma de este modelo, no fue posible hallar muerte neuronal en la AMeXM (fig. 5). Si bien se utilizaron animales con sobrevidas de hasta 24h, se emplearon también animales con sobrevidas de hasta 30 días tras la inyección de PTZ y en ningún caso se halló signo alguno de muerte neuronal. Es de destacar, sin embargo, que en el hipocampo de animales con sobrevidas de 24h fue posible hallar signos de neurodegeneración de terminales axónicos en el área CA3 (resultados no mostrados). Esta característica no se observó en otra estructura cerebral.
 que muestran en A el núcleo amigdalino medial posterodorsal y el posteroventral, en B el núcleo intersticial de la estría terminal anterior y ventral de animales con sobrevida de 24h luego del estado epiléptico y que se procesó con la técnica A-Cu-Ag.")
Fotografías de secciones de cerebros (×2,5) que muestran en A el núcleo amigdalino medial posterodorsal y el posteroventral, en B el núcleo intersticial de la estría terminal anterior y ventral de animales con sobrevida de 24h luego del estado epiléptico y que se procesó con la técnica A-Cu-Ag.
Numerosos hallazgos ponen de manifiesto el hecho de que el sistema gabaérgico se encuentra involucrado en diferentes tipos de epilepsias. Por ejemplo, Muñoz et al19, analizando cerebros de pacientes epilépticos, hallaron una reducción de inmunotinción para el receptor GABAb 1a-b en la capa de células granulares del giro dentado del hipocampo. Otros autores encontraron una sobreexpresión de los dos subtipos que presenta el receptor GABAB (1 y 2) en el hipocampo de pacientes con epilepsia del lóbulo temporal, que se interpreta como un mecanismo compensatorio20. También se encontraron alterados los valores del receptor GABAA en cerebros de pacientes epilépticos y en modelos animales21.
Así, el GABA se encuentra implicado en la epilepsia, el estado epiléptico y otros síndromes epilépticos. Alteraciones postsinápticas y presinápticas, como cambios en la composición de subunidades de receptores, estructura de los receptores o modificaciones genéticas del receptor, podrían afectar a la susceptibilidad a una crisis epiléptica.
No obstante, la epilepsia no es un síndrome gabaérgico. Diversos hallazgos llevaron a la idea de que las crisis epilépticas responderían a un modelo simple en el que la inhibición y la excitación actuarían como freno y acelerador, respectivamente. Sin embargo, este esquema es un modelo demasiado simplificado. En primer lugar, hay datos que indican que la neurotransmisión gabérgica puede ser excitadora en condiciones basales no sólo en el cerebro en desarrollo22, sino también en el cerebro epiléptico23.
En resumen, en cerebros epilépticos humanos y en modelos animales se ha encontrado un aumento en el número de sinapsis gabaérgicas; esto se interpreta como mecanismos compensatorios, aunque también podrían explicarse como secundarios a cambios en la función gabaérgica (de inhibición a excitación) y que serían en parte los orígenes de la hiperexcitabilidad del sistema1.
Expresión de genes tempranos y epilepsiaEstudios previos han utilizado la proteína fos para valorar la participación de distintas poblaciones neuronales en modelos de epilepsia usando PTZ24,25. Aquí se demuestra, luego del análisis cuantitativo de núcleos de neuronas activadas con fos, que la expresión de ésta es máxima a las 2 y 4h de sobrevida consistente con la expresión aguda, aunque se evidencia una expresión sostenida hasta las 12h luego del EE. A partir de las 12h, la inmunomarcación disminuye considerablemente, por lo que en este estudio se decidió no sacrificar animales con más de 24h de sobrevida luego de la inyección.
Tomando la expresión de la proteína fos como una herramienta de cartografía funcional y anatómica, se muestra aquí la capacidad de la AMeXM de reaccionar activándose a través de la expresión de genes tempranos, como c-fos, y así su participación en mecanismos epilépticos. Ello permite afirmar que la AMeXM se encuentra involucrada en procesos epilépticos que implican la participación de mecanismos gabaérgicos.
El hecho de que la expresión de fos fuera evidente a las 24h apuntó a la hipótesis de que estas neuronas podrían estar sufriendo algún proceso neurodegenerativo. Para ello, se procesaron cortes paralelos de cerebros con la técnica A-Cu-Ag. A pesar de utilizar diferentes dosis de PTZ (desde convulsiones leves a un EE severo), no se encontró degeneración en estos cerebros, lo que pone de manifiesto que, si bien la AMeXM se encuentra involucrada en este modelo animal, sus neuronas fueron capaces de recuperarse del estrés que significa el EE inducido por PTZ.
La participación de la amígdala extendida medial en mecanismos epilépticosEl complejo amigdalino ha sido poco estudiado en el campo de la epilepsia, a pesar de haberse demostrado su implicación, principalmente del lóbulo temporal26.
En investigación básica hay estudios que abordan el problema de la epilepsia con distintos modelos animales. En ellos se pone énfasis en el hipocampo y en algunos núcleos amigdalinos del complejo basolateral, y dejan de lado los otros complejos amigdalinos. Teniendo en cuenta lo anterior, la pregunta que surge es: ¿se encuentra realmente involucrada la AMeXM en la epilepsia? Los resultados aquí obtenidos permiten afirmar una clara participación de la AMeXM en mecanismos epilépticos.
Si bien al utilizar el producto de genes tempranos como c-fos como un marcador de la actividad neural se observa que otras áreas cerebrales también son activadas, aquí se deben considerar dos puntos importantes:
- 1.
El modelo animal aquí utilizado corresponde a una epilepsia secundariamente generalizada y, por lo tanto, se activarán además otras áreas cerebrales.
- 2.
En estas áreas la expresión de fos disminuye rápidamente (a las 4h), mientras que en las estructuras de la AMeXM la expresión de fos se sostiene significativamente hasta las 12h de sobrevida.
Los resultados obtenidos aquí demuestran la participación de la AMeXM en mecanismos epilépticos pero, a diferencia de lo que ocurre en otros modelos animales como, por ejemplo, ácido kaínico27 o pilocarpina28, no se halló muerte neuronal en la AMeXM. Hay informes de que la inyección sistémica de PTZ induce neurodegeneración en neuronas del hipocampo y la amígdala29; sin embargo, en estos estudios la neurodegeneración hallada es de tipo apoptótica, la cual no puede detectarse con la técnica utilizada en este trabajo. Además, en este estudio se considera la amígdala como un complejo unitario; no discrimina las distintas subdivisiones. Asimismo, se ha demostrado que el EE inducido por PTZ induce estrés oxidativo en el cerebro de ratas, con la consecuente muerte de neuronas30. La técnica aquí utilizada es específica para la detección de muerte neuronal por necrosis y muy probablemente la muerte de neuronas inducida por estrés oxidativo involucre un componente apoptótico31.
Otros estudios16 han mostrado que un estado epiléptico inducido con PTZ en ratas inmaduras causa un periodo de fuerte activación caracterizada por un incremento agudo de la tasa metabólica, lo cual tiene como consecuencia, en la madurez, cambios permanentes en la permeabilidad de la membrana celular, permitiendo la entrada de fucsina ácida. Sin embargo, estas células nunca entran en la fase de la fragmentación del ADN y son capaces de recuperarse.
Los resultados contradictorios se deben quizá a la utilización de distintas técnicas para la detección de la muerte neuronal o por considerar el complejo amigdalino como una totalidad sin tener en cuenta sus diferentes subdivisiones. Nuestros hallazgos demuestran que el PTZ, antagonista gabaérgico, no causa muerte neuronal en la AMeXM, lo que pone de manifiesto que, en las epilepsias en que la causa subyacente es mayormente un componente inhibitorio, probablemente no tengan como consecuencia la muerte de sus neuronas.
Proyecciones clínicas y prácticas de la investigaciónSi bien el presente trabajo tiene una orientación netamente básica, de él pueden extraerse importantes conclusiones. En primer lugar, esta investigación constituye una base para comprender la complejidad del complejo amigdalino. Éste es una estructura cerebral que, aunque su tamaño es menor, no lo es su complejidad, ya sea en sus funciones y su anatomía, en términos de sus conexiones con otras estructuras.
Este trabajo es el punto de partida para nuevas investigaciones tanto básicas como clínicas. En lo básico es importante para el desarrollo de nuevos fármacos antiepilépticos antagonistas o agonistas gabaérgicos que interfieran con el proceso epileptógeno; en el aspecto clínico, estableciendo una correlación de lo aquí trabajado con la epilepsia del lóbulo temporal resistente y en la elección de diferentes fármacos para su tratamiento.
El conocimiento detallado de la anatomía de la amígdala permitirá, en un futuro próximo, realizar una selectiva cirugía de la epilepsia del lóbulo temporal. Todo ello enmarcado en un esfuerzo de la investigación básica en interrelación constante con la clínica para aportar nuevas iniciativas para comprender con mayor profundidad esta enfermedad.
FinanciaciónEste trabajo ha recibido financiación de la Secretaría de Ciencia y Tecnología de la Universidad Nacional de Córdoba.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

