



La gliomatosis cerebri (GC) es un tumor glial difuso infrecuente, caracterizado por una gran capacidad infiltrativa. La variabilidad de características histológicas y de grado que presenta, junto con respuestas generalmente pobres a los tratamientos, convierte a la GC en un tumor poco conocido con diversas cuestiones por responder. En este volumen de Neurología se presenta un estudio sobre las características clínicas, evolución y respuestas al tratamiento en una serie de 22 pacientes con GC.
DesarrolloLa GC presenta una clínica poco distintiva, junto con una neuroimagen característica pero poco específica, lo que implica la realización de un diagnóstico diferencial amplio. Aunque en su evolución parecen estar implicados factores pronósticos similares a los de los otros gliomas, la heterogeneidad de sus hallazgos patológicos y moleculares dificulta su precisa caracterización. Así se explican sus variables comportamiento y respuesta al tratamiento. La evidencia sobre la eficacia de distintos abordajes terapéuticos se basa en series de casos clínicos; por lo tanto, el tratamiento no está bien establecido. Parte de la investigación actual se centra en identificar factores moleculares que puedan ser predictores de la respuesta a la quimioterapia.
ConclusionesLa incorporación de quimioterapia al tratamiento clásico basado en la radioterapia parece ofrecer mayores tasas de respuestas, aunque su impacto en la supervivencia sigue siendo escaso. Es necesaria la realización de estudios multicéntricos de fase III para evaluar las diferentes estrategias terapéuticas. Asimismo, hay que profundizar en el conocimiento sobre la histogénesis y los factores moleculares pronóstico para poder estratificar adecuadamente a los pacientes.
Gliomatosis cerebri (GC) is a rare, diffusely growing glial tumour characterized by extensive brain infiltration. The diversity of histological subtype and grade on presentation among different subjects, in addition to the usually poor response to treatment make GC an uncertain entity where many questions still remain unanswered. One article in this issue of Neurología describes a series of 22 patients with GC, where clinical, therapeutic and outcome results are detailed.
DevelopmentClinical presentation of GC is non-specific and, although the neuroimage is characteristic, the spectrum of differential diagnosis is wide. Despite the fact that known prognostic factors in glioma also seem to be involved in GC, the heterogeneity of pathology and molecular findings on biopsy samples makes it difficult to characterise GC correctly. Therefore, variability of outcome and response to therapy is the rule. Evidence on therapeutic strategies is based on case-series. According to this, the optimal treatment is not well established. Part of current research is focused on identifying molecular predictor factors of response to chemotherapy.
ConclusionsThe addition of chemotherapy in the classic treatment schedule based on radiotherapy seems to produce better responses in GC patients. However, the outcome of these patients remains poor with low survival rates. Phase III multi-centre trials to evaluate different therapeutic strategies in GC are essential. Further knowledge on the histological profile and molecular prognostic factors is also required. Patients should be stratified according to the prognostic factors identified.
La gliomatosis cerebri (GC) es un tumor cerebral primario infrecuente, probablemente representa menos del 1% de todos los astrocitomas1,2, con una histogénesis y una historia natural intrigantes aún por definir. Además, la GC presenta una pobre y discordante respuesta a los tratamientos. En este volumen de Neurología se presenta un artículo sobre una serie de pacientes con GC que contribuye a mejorar nuestro conocimiento sobre la evolución y la respuesta al tratamiento de esta problemática entidad.
DefiniciónDe acuerdo con los criterios de la World Health Organization (WHO) Classification of Tumours, la GC es un glioma difuso, con un patrón de crecimiento excepcionalmente infiltrativo y extenso que suele preservar la arquitectura de los tejidos neurales circundantes. Debe afectar a un mínimo de 3 lóbulos cerebrales y puede infiltrar sustancia gris tanto cortical como de ganglios basales, así como extenderse a lo largo del tronco cerebral y la médula espinal2. El fenotipo celular habitualmente es de tipo astrocitario aunque también admite el oligodendrocitario y el oligoastrocitario3. Se ha dividido la GC en tipo 1, cuando no se observa una masa tumoral en el patrón infiltrativo, y en tipo 2, cuando además de la infiltración cerebral se objetiva una lesión con efecto de masa, habitualmente pequeña (< 1cm de diámetro). Cuando otros tipos de gliomas presentan una extensa progresión de características infiltrativas, más que crecimiento de la masa tumoral en sí misma, se denomina a estos fenómenos GC secundaria2,4,5.
La GC presenta un comportamiento biológico habitualmente agresivo, por lo que la WHO la clasifica como grado III de malignidad. Debido a que la mayoría de las muestras patológicas consisten en biopsias por la irresecabilidad que conlleva su extensión, éstas pueden no ser representativas de la totalidad del tumor. Por este motivo, aunque haya ausencia de rasgos correspondientes a anaplasia celular, sigue considerándose como una tumoración de alto grado.
Epidemiología, clínica y neuroimagenLa incidencia de la GC es más elevada en varones que en mujeres (1,3:1), con una tasa probable de alrededor 0,6-8,2 casos/año4,6. Aunque el intervalo de edad al diagnóstico va desde los primeros meses de vida hasta la senectud, la mayoría de los pacientes tienen entre 40 y 50 años4. La presentación clínica es variable y no es una ayuda para el diagnóstico; habitualmente, son los cambios no específicos de la resonancia magnética (RM) lo que despertará la sospecha diagnóstica. En ésta, además de observarse extensos cambios de señal hiperintensos en las secuencias FLAIR y T2, no objetivables en las T1 o en la tomografía computarizada, pueden evidenciarse algunas alteraciones indicativas, pero no específicas, de GC. Éstas consistirían en un engrosamiento del cuerpo calloso, ligeros signos de colapso de astas ventriculares y tumefacción hemisféricas, pérdida de diferenciación entre sustancia blanca y gris, engrasamiento del córtex o ganglios basales y, habitualmente, preservación del cerebelo aunque haya afectación troncoencefálica. Puede observarse también una captación de contraste que puede ser desde difusa y poco uniforme a muy evidente, presente hasta en la mitad de los pacientes; este hallazgo es más frecuente en las GC tipo 27–9. La falta de especificidad de la neuroimagen y de la clínica plantea el diagnóstico diferencial con el síndrome de Behçet, el de Sjögren, la leucoencefalopatía isquémica, posradioterápica multifocal progresiva o posterior reversible, las encefalitis infecciosas o inmunitarias, las leucodistrofias, vasculitis, enfermedades desmielinizantes, ciertas formas de linfomas cerebrales primarios o algunos tipos de ictus como el CADASIL; la valoración de la RM, junto con el adecuado contexto clínico, es lo que permitirá determinar la indicación de la biopsia y confirmar el diagnóstico. La espectroscopia y las secuencias de volumen sanguíneo cerebral relativo pueden ser de gran ayuda para orientar las lesiones reflejadas en la RM como tumorales de estirpe glial, así como servir de guía para orientar hacia la zona donde realizar la biopsia10–12.
Alteraciones genéticas y molecularesLa GC tanto por su extensión como por el hecho de presentar heterogeneidad en el grado y el subtipo histológico, así como la ausencia de un hallazgo histopatológico claramente específico, plantea diversas incógnitas sobre su histogénesis. Por una parte, puede tratarse simplemente de un subtipo de glioma difuso con una excepcional capacidad infiltrativa que, al adquirir una serie de cambios moleculares, similares a los que suceden en las transformaciones de astrocitomas de bajo grado a anaplásicos o glioblastomas secundarios, malignizaría y probablemente se observaría como GC de tipo 2. Esta hipótesis estaría apoyada por la presencia de mutaciones en p53, PTEN o por la amplificación del EGFR. Estas alteraciones se han encontrado en la GC, pero con una frecuencia mucho menor que la de los astrocitomas convencionales13–19. También se han descrito otras alteraciones en relación con los mecanismos de control del ciclo celular, como la disminución de la expresión de p27 en relación con la progresión de la GC14–16, lo que apoyaría la evolución de una tumoración de bajo a alto grado. Por otro lado, una hipótesis alternativa defendería que la GC es una entidad independiente del glioma difuso. Sus argumentos se basan en el hallazgo de alteraciones cromosómicas y marcadores moleculares distintos de los de los astrocitomas20. De forma interesante, algunos trabajos han identificado en muestras de GC un elevado porcentaje de células positivas para Sox2 y Nestin, marcadores de precursores neurales21,22, y para CD34, marcador de stem cells22, lo que reflejaría una naturaleza más primitiva de la GC respecto a los astrocitomas. Únicamente existe más consenso en afirmar que la GC es un tumor de origen monoclonal, con lo que se descarta la posibilidad de una transformación neoplásica de distintas áreas cerebrales que acaban confluyendo14,16. La principal causa de la disparidad de hallazgos e interpretaciones es la escasez de buen material anatomopatológico para realizar los adecuados análisis moleculares, ya que en la mayoría de los casos se trata de material procedente de biopsia con alta presencia de tejido cerebral normal.
Factores pronósticosEn cuanto a la evolución de la GC, los factores independientes de buen pronóstico más firmemente establecidos en relación con la supervivencia son, al igual que en otros gliomas, el índice de Karnofsky (≥ 70-80) y el grado histológico de la muestra analizada4,7. La edad como factor pronóstico presenta, en la mayoría de los estudios, una tendencia hacia la significación, aunque sin alcanzarla; los pacientes jóvenes presentan supervivencias discretamente más prolongadas4,7,9. Otros factores de buen pronóstico a considerar son el subtipo histológico (oligodendrogliomas mejor que el resto)4, la ausencia de captación de contraste (probablemente relacionada con el grado histológico)6,9 y una menor infiltración de sustancia gris respecto a la blanca12. Estudios aún no reproducidos indican que las deleciones de los cromosomas 13q y 10q y ganancias del 7q, así como mutaciones en p53 y PTEN, se correlacionan con una pobre supervivencia9,15. En el campo molecular, en una serie donde predomina el tipo histológico oligodendrocitario, se observa que las deleciones 1p19q se asocian a una mayor supervivencia, como ocurre con los oligodendrogliomas23. Además, existe una correlación significativa entre la presencia de esta mutación y la metilación de la MGMT, aunque en estos pacientes la ausencia de la metilación se asocia únicamente a una tendencia no significativa hacia un menor intervalo libre de progresión tumoral8.
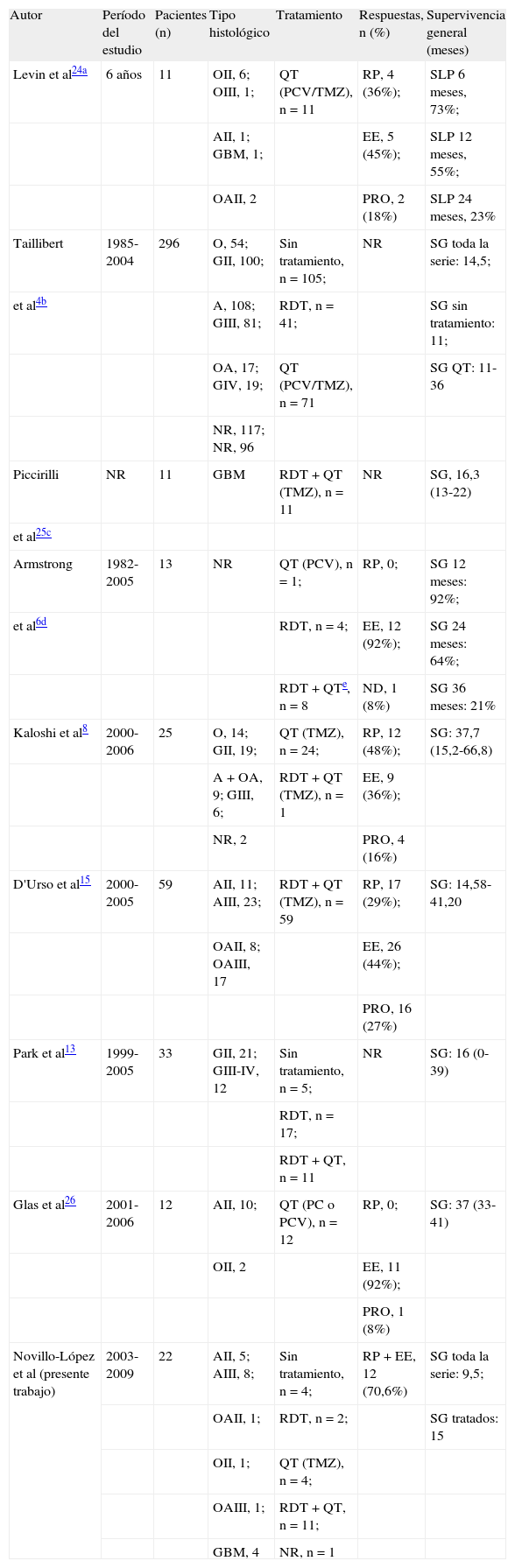
TratamientoEl esquema de tratamiento de la GC aún no está definido. La GC es una tumoración irresecable, por lo que la cirugía sólo debe tener un papel diagnóstico. La única evidencia sobre la eficacia del tratamiento oncológico, quimioterapia (QT) y/o radioterapia (RDT), proviene de series de casos clínicos o revisiones retrospectivas de casos aislados (tabla 1)4,6,8,13,24–26. Al igual que en los demás gliomas, la RDT ha constituido la base del tratamiento de la GC hasta la última década. Su impacto relativo y contradictorio en la supervivencia, la falta de consenso en la dosis total, dosis/fracción y el tipo de irradiación (focal u holocraneal), así como los potenciales efectos adversos generados al irradiar grandes campos cerebrales, no despejan las dudas sobre su beneficio terapéutico2,4,7. La administración de QT, junto con la RDT o de forma aislada, es una práctica cada vez más extendida, aunque también carece de un nivel de evidencia alto que lo respalde. Los quimioterápicos habitualmente empleados son esquemas de PCV (procarbacina, lomustina, vincristina) y temozolomida (TMZ), sin diferencias significativas en cuanto a supervivencia entre ambos, aunque la TMZ presenta un mejor perfil de efectos adversos y permite administraciones prolongadas en el tiempo4,5,8,24–26. La administración aislada de QT, sobre todo con TMZ, ha irrumpido como posible estrategia de primera línea en la GC de estirpe oligodendroglial o en tumores que presenten la deleción 1p19q4,23, con lo que la RDT queda como tratamiento de rescate después de la progresión a la QT. Esto permitiría retrasar la toxicidad tras radiación manteniendo cierta tasa de respuestas a esta segunda línea de tratamiento. Obviamente, si la estirpe histológica de la GC corresponde a un
Resumen de los estudios más recientes sobre tratamiento en gliomatosis cerebri
| Autor | Período del estudio | Pacientes (n) | Tipo histológico | Tratamiento | Respuestas, n (%) | Supervivencia general (meses) |
| Levin et al24a | 6 años | 11 | OII, 6; OIII, 1; | QT (PCV/TMZ), n = 11 | RP, 4 (36%); | SLP 6 meses, 73%; |
| AII, 1; GBM, 1; | EE, 5 (45%); | SLP 12 meses, 55%; | ||||
| OAII, 2 | PRO, 2 (18%) | SLP 24 meses, 23% | ||||
| Taillibert | 1985-2004 | 296 | O, 54; GII, 100; | Sin tratamiento, n = 105; | NR | SG toda la serie: 14,5; |
| et al4b | A, 108; GIII, 81; | RDT, n = 41; | SG sin tratamiento: 11; | |||
| OA, 17; GIV, 19; | QT (PCV/TMZ), n = 71 | SG QT: 11-36 | ||||
| NR, 117; NR, 96 | ||||||
| Piccirilli | NR | 11 | GBM | RDT + QT (TMZ), n = 11 | NR | SG, 16,3 (13-22) |
| et al25c | ||||||
| Armstrong | 1982-2005 | 13 | NR | QT (PCV), n = 1; | RP, 0; | SG 12 meses: 92%; |
| et al6d | RDT, n = 4; | EE, 12 (92%); | SG 24 meses: 64%; | |||
| RDT + QTe, n = 8 | ND, 1 (8%) | SG 36 meses: 21% | ||||
| Kaloshi et al8 | 2000-2006 | 25 | O, 14; GII, 19; | QT (TMZ), n = 24; | RP, 12 (48%); | SG: 37,7 (15,2-66,8) |
| A + OA, 9; GIII, 6; | RDT + QT (TMZ), n = 1 | EE, 9 (36%); | ||||
| NR, 2 | PRO, 4 (16%) | |||||
| D'Urso et al15 | 2000-2005 | 59 | AII, 11; AIII, 23; | RDT + QT (TMZ), n = 59 | RP, 17 (29%); | SG: 14,58-41,20 |
| OAII, 8; OAIII, 17 | EE, 26 (44%); | |||||
| PRO, 16 (27%) | ||||||
| Park et al13 | 1999-2005 | 33 | GII, 21; GIII-IV, 12 | Sin tratamiento, n = 5; | NR | SG: 16 (0-39) |
| RDT, n = 17; | ||||||
| RDT + QT, n = 11 | ||||||
| Glas et al26 | 2001-2006 | 12 | AII, 10; | QT (PC o PCV), n = 12 | RP, 0; | SG: 37 (33-41) |
| OII, 2 | EE, 11 (92%); | |||||
| PRO, 1 (8%) | ||||||
| Novillo-López et al (presente trabajo) | 2003-2009 | 22 | AII, 5; AIII, 8; | Sin tratamiento, n =4; | RP + EE, 12 (70,6%) | SG toda la serie: 9,5; |
| OAII, 1; | RDT, n = 2; | SG tratados: 15 | ||||
| OII, 1; | QT (TMZ), n = 4; | |||||
| OAIII, 1; | RDT + QT, n = 11; | |||||
| GBM, 4 | NR, n = 1 |
A: astrocitoma; EE: enfermedad estable; GBM: glioblastoma; GII: grado 2; GIII: grado 3; MTX: metotrexato; NR: no comunicado; OA: oligoastrocitoma; O: oligodendroglioma; PC: procarbacina + lomustina; PCV: procarbacina + lomustina + vincristina; PRO: progresión; QT: quimioterapia; RDT: radioterapia; RP: respuesta parcial (incluye respuesta menor); SG: supervivencia general; SLP: supervivencia libre de progresión; TMZ: temozolomida; VCR: vincristina.
glioblastoma, el tratamiento recomendado sería el régimen de Stupp27. Por otro lado, la utilidad del empleo de agentes antiangiogénicos (bevacizumab, celecoxib) en la GC está aún por establecer28. Finalmente, cabe resaltar la escasa utilidad de los corticoides en la GC, ya que existe un reducido componente de edema vasogénico, sobre todo en GC de tipo 1.
ConclusionesEl estudio presentado por Novillo-López et al pone de manifiesto la heterogeneidad histológica de este tumor y su aparente respuesta al tratamiento, aunque desgraciadamente con una evolución superponible a la de los glioblastomas, incluso en la tendencia a producir fenómenos trombóticos. La dificultad diagnóstica en vida hasta la aparición de la RM y el predominio de material patológico de biopsias han dificultado la caracterización de la GC. Este hecho se refleja en que es el único tumor cerebral que, para sus criterios diagnósticos, requiere indispensablemente de la neuroimagen y hasta se subclasifica en función de ésta, lo que forzosamente no se correlaciona con su conducta biológica. Una conclusión a que nos conduce la heterogeneidad de los hallazgos histológicos y moleculares, así como de su historia natural, es que nos encontramos ante un tumor de origen indeterminado, con una inestabilidad genómica aún por identificar que le facilita, en comparación con los astrocitomas clásicos, una rápida progresión por distintos grados de malignidad (III y IV). Estas circunstancias explicarían las divergencias en su supervivencia y en las respuestas a los tratamientos, por lo que sería aconsejable, en futuros estudios, separar a los pacientes con GC tipo 2 o con histología de glioblastoma de los demás. Asimismo, esta entidad pone de manifiesto que la clasificación anatomopatológica clásica para determinar la agresividad de los tumores es limitada; se necesita identificar “firmas” genéticas o moleculares propias que determinen con mayor precisión la evolución de los distintos tumores, esto facilitaría y hasta personalizaría la homogeneidad de los ensayos y los tratamientos. Profundizar en el conocimiento de la GC no sólo beneficiará a estos enfermos, sino que probablemente ayudará a comprender los mecanismos de migración infiltrativa de las gliomatosis secundarias, hoy bastante frecuentes como resultado del uso de antiangiogénicos en el tratamiento de los glioblastomas.

