



La encefalopatía por glicina (GCE. MIM 605899) o hiperglicinemia no cetósica (NKH) es un error innato del metabolismo (EIM) con herencia autosómica recesiva1, donde se afecta el sistema mitocondrial de clivaje de la glicina2. En consecuencia, se acumula glicina, que estimula los receptores N-metil-D-aspartato (NMDA) y ocasiona la mayoría de los síntomas3. Se describen varias formas clínicas de la enfermedad; de ellas, la forma clásica es la más frecuente y se caracteriza por hipotonía, disminución del reflejo moro, convulsiones mioclónicas, apnea, letargia y coma; los síntomas aparecen en los primeros días de vida4. Se describen otras variantes menos frecuentes: la forma de presentación tardía5, 3 variantes atípicas con síntomas más heterogéneos1,6.
En esta enfermedad el electroencefalograma (EEG) muestra un trazado de estallido/supresión durante el primer mes de vida4, que cambia a hipsarritmia. El diagnóstico bioquímico consiste en la cuantificación de glicina en plasma/suero y LCR; y el análisis simultáneo de ácidos orgánicos en orina para descartar una aciduria orgánica3. La relación de niveles de glicina en LCR/plasma es superior a 0,08 en la variante clásica, mientras que en las variantes atípicas se encuentra entre 0,04-0,023,5. Sin embargo, otros autores no han encontrado correspondencia entre los síntomas y la relación de glicina en LCR/plasma7,8. El tratamiento se basa en la restricción proteica moderada en combinación con benzoato de sodio. También se pueden administrar antagonistas del receptor NMDA9, mientras que el tratamiento con valproato está contraindicado, pues eleva los niveles de glicina10,11.
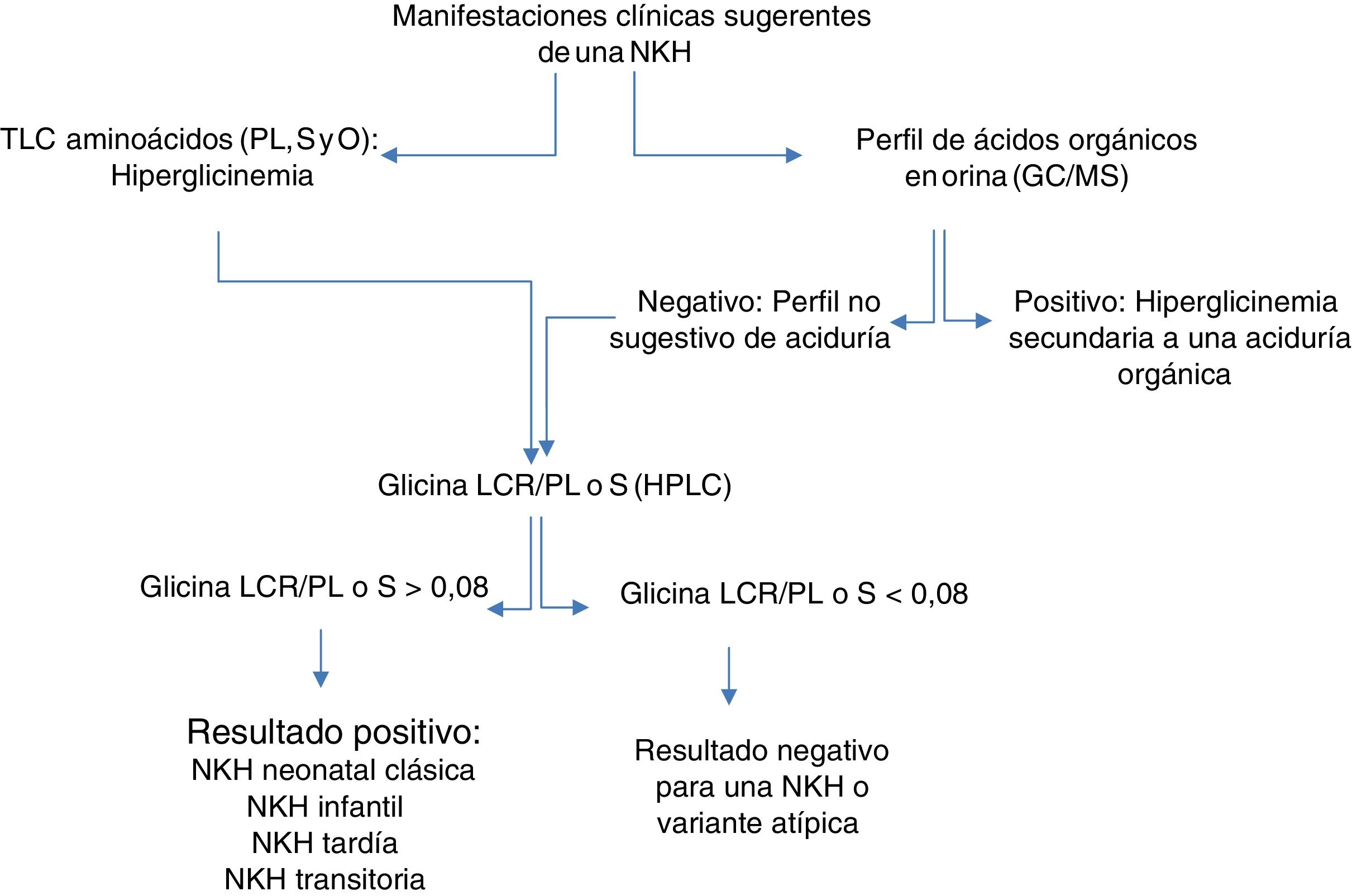
En Cuba, suele sospecharse una aciduria orgánica en los pacientes con HKH. Por tanto, se debe descartar una NKH en pacientes con hipotonía, encefalopatía y convulsiones; donde no se haya diagnosticado un EIM. Describimos los hallazgos clínicos y bioquímicos en 2 pacientes con sospecha clínica de NKH, así como el algoritmo empleado para el diagnóstico bioquímico (fig. 1).

Protocolo para el diagnóstico bioquímico de la hiperglicinemia no cetósica.
C-MS: cromatografía de gases acoplado a espectrometría de masa; GLCR: líquido cefalorraquídeo; HPLC: cromatografía líquida de alta resolución; O: orina; PL: plasma; S: suero; TLC: cromatografía en placa delgada.
Paciente 1
Comenzó, a los 28 días de nacido, con un cuadro neurológico; hipotonía, letargia, apnea, convulsiones tipo mioclonías, sospechándose de un síndrome de West, el cual se confirma por clínica y EGG con patrón de estallido-supresión en las primeras semanas e hipsarritmia a partir del mes de su nacimiento. Al inicio llevó tratamiento con valproato y gabapentina (vigabatrina 60mg/kg/día), pero no se logró mejoría. Se inicia tratamiento con ACTH y topiramato, lográndose resolver los episodios convulsivos. La cuantificación de glicina mostró niveles en LCR de 109uM, suero 351uM y una relación LCR/suero de 0,3 (normal: <0,08). Al mes se recibieron muestras de la paciente obteniéndose niveles de glicina en suero de 366uM, en LCR de 130uM y una relación de 0,4, diagnosticándose una NKH neonatal clásica.
Paciente 2
Comenzó con manifestaciones clínicas sugerentes de un EIM al año de edad. Los principales síntomas consistieron en epilepsia generalizada de difícil control, retraso mental, retraso motor y EGG patológico. El paciente se trató con valproato de sodio (750mg/día) y clobazam (25mg/día). La cuantificación de glicina mostró niveles en LCR de 102uM, suero 88uM y una relación LCR/suero de 1,2. Debido a los resultados obtenidos y al cuadro clínico, se diagnostica una NKH del tipo infantil o asociada a terapia con valproato. Al repetir la cuantificación de glicina, una vez suspendido el tratamiento con valproato, los niveles de glicina disminuyeron a 38uM y 112uM en LCR y suero, respectivamente. Sin embargo, la relación LCR/suero fue de 0,3, diagnosticándose una NKH infantil.
El diagnóstico bioquímico de la NKH en Cuba se realiza en pacientes sintomáticos, pues no existe un programa de pesquisa neonatal para esta enfermedad. La cuantificación de glicina en LCR debe sugerirse en casos con niveles aumentados de glicina en orina y sangre, donde se haya descartado una aciduria orgánica. El asesoramiento genético a padres y familiares de los pacientes afectados se limitó a informar el riesgo de recurrencia del 25%, por tratarse de un defecto autosómico recesivo. Asimismo, el pronóstico se predijo a partir de la edad y los síntomas de comienzo, al no disponer de la caracterización molecular.
La determinación de la relación glicina LCR/suero permitirá diagnosticar la variante clásica y la mayoría de las variantes atípicas1,5,12,13. No obstante, se recomienda implementar alternativas para confirmar el diagnóstico de NKH en los pacientes con resultados positivos o en aquellos que pueden tener una variante atípica de la enfermedad con una relación glicina LCR/suero normal.

