



El hipopituitarismo es un síndrome clínico, resultante de la secreción insuficiente o ausencia completa de secreción de una o varias hormonas hipofisarias anteriores. Aunque típicamente el diagnóstico se suele realizar en período neonatal, en ocasiones, su primera manifestación puede ser un retraso psicomotor en loa lactantes, por lo que el diagnóstico precoz puede prevenir el deterioro neurocognitivo, evitando la aparición de secuelas neurológicas1.
Se presentan 2 casos de hipopituitarismo, diagnosticados a raíz de la presencia de retraso psicomotor en periodo de lactante:
Caso 1Recién nacida mujer a término, controlada por aumento craneal del diámetro biparietal y ventriculomegalia de ventrículos laterales con colpocefalia, sin signos de hipertensión intracraneal. Con 20 meses persiste ausencia de bipedestación y lenguaje; estudio analítico neurometabólico, carga viral de citomegalovirus, electroencefalograma, potenciales evocados auditivos, fondo de ojo, cariotipo y cribado neonatal normal. A los 21 meses presenta longitud de 70,3cm (–4,8DE) y peso de 6,9kg (–3,71 DE), baja velocidad de crecimiento y estudio analítico hormonal compatible con déficit de hormona de crecimiento (GH), con el resto de estudios, incluido el tiroideo, normal. En el estudio genético arrays-CGH se detecta una deleción 1q25.2, asociado a haploinsuficiencia del gen LHX4, implicado en el hipopituitarismo. A los 23 meses de edad presenta T4 libre 0,51ng/dl (VN: 0,58-1,64), con TSH 0,97μUI/ml (VN: 0,34-5,6), anormalmente normal para el nivel de T4 libre. Resto del eje hipotálamo-hipofisario normal (glucemia, iones, ACTH, cortisol, IGF1, IGFBP3 y prolactina). Ante hipotiroidismo central, se inicia tratamiento con levotiroxina (25μg/día/vía oral) con clara mejoría del tono muscular y desarrollo psicomotor. En la resonancia magnética nuclear (RMN), se aprecia aumento del sistema ventricular lateral bilateral con colpocefalia, con hipoplasia hipofisaria en situación anatómica.
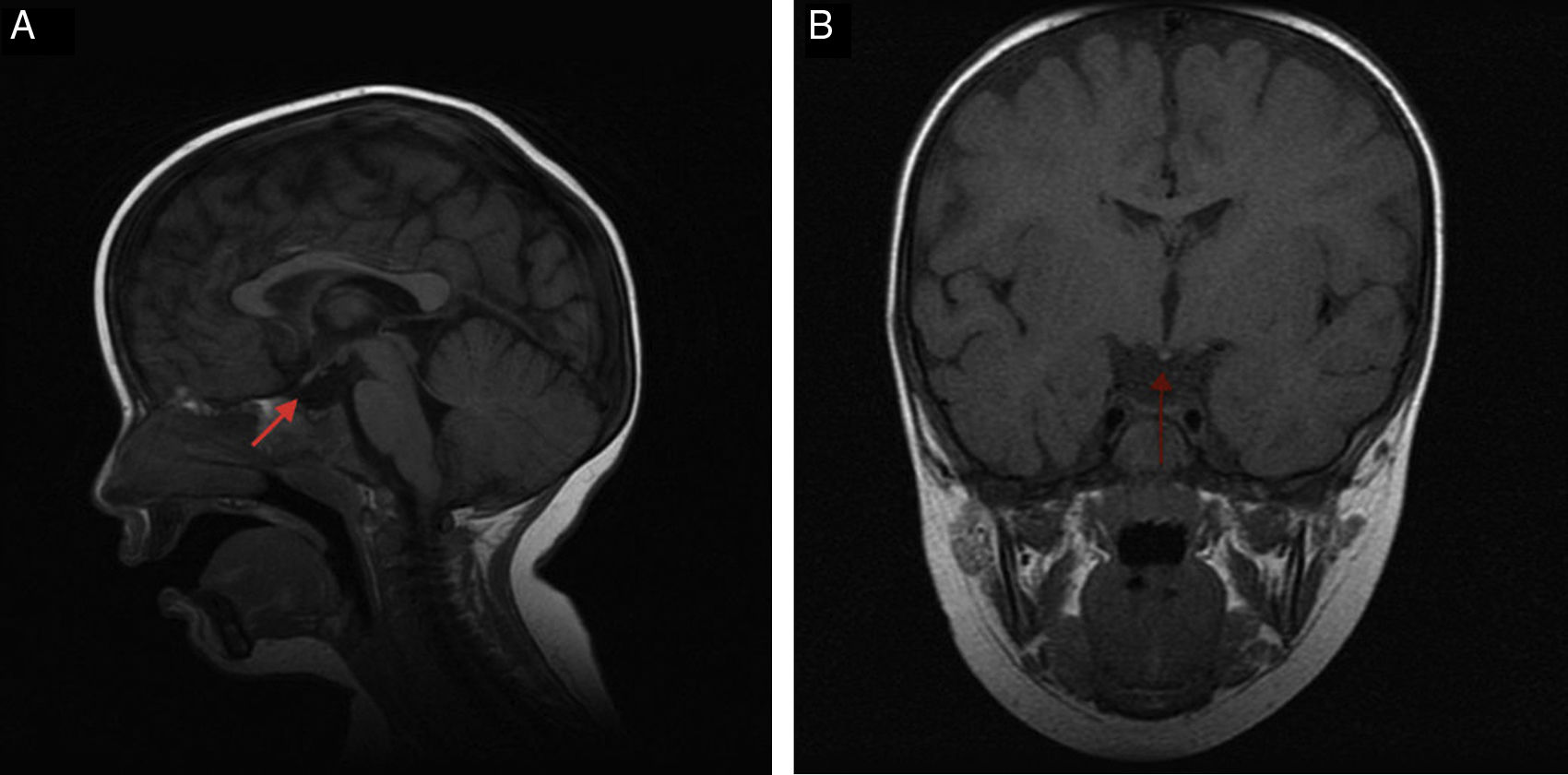
Caso 2Recién nacido varón a término, con antecedente de hipoglucemia sintomática al nacimiento, ictericia mucocutánea que precisó fototerapia, escasa succión e hipotonía cervicoaxial llamativa, con estudio en período neonatal normal. En la determinación analítica a los 12 meses de edad por persistencia de hipotonía, ausencia de sedestación y estreñimiento persistente, presenta T4 libre 0,50ng/dl (VN: 0,58-1,64), con TSH 2,34μUI/ml (VN: 0,34-5,6). Resto del eje hipotálamo hipofisario normal (glucemia, ACTH, cortisol, LH, FSH, testosterona, prolactina, IGF1 e IGFBP3). Ante hipotiroidismo central se inicia tratamiento con levotiroxina (25μg/día/vía oral) presentando mejoría del estreñimiento y del desarrollo psicomotor. Presenta escroto hipoplásico, testes en ascensor y longitud del pene en el límite bajo de la normalidad. A los 2 años, refiere cuadro de astenia importante tras juegos, somnolencia con torpeza y caídas frecuentes, presentando un cortisol 2,71μg/dl (VN: 5-25) con ACTH 7,7pg/ml (VN: 5-46) e iones normales, por lo que se inicia tratamiento sustitutivo con hidrocortisona con mejoría clínica. En la RMN presenta hipoplasia moderada de adenohipófisis, neurohipófisis ectópica localizada en la eminencia media y agenesia completa del tallo hipofisario (fig. 1A y B). Estudio genético negativo.
 y coronal (B). Hipoplasia moderada de adenohipófisis, neurohipófisis ectópica en la eminencia media y agenesia completa del tallo hipofisario.")
La etiología del hipopituitarismo es variable, existiendo causas adquiridas (tumores seguido de malformaciones del sistema nervioso central)2, genéticas (alteraciones en los factores de transcripción: PIT1, PROP1, HESX1, LHX3, LHX4, PITX1, PITX2, TPIT y SOX3)3 e idiopáticas.
El diagnóstico de sospecha es clínico, con una expresión variable y heterogénea en función de la severidad y del número de hormonas alteradas, y de la rapidez de inicio del cuadro. Lo más típico en periodo neonatal es la hipoglucemia, ictericia prolongada con colestasis, micropene, alteración de la línea media, clínica neurológica o retraso psicomotor con hipotonía, letargia o alteración de la succión4. Es fundamental sospecharlo para conseguir un diagnóstico lo más precoz posible, que limite el retraso neurocognitivo que suele acompañar a esta enfermedad5.
El diagnóstico se establece basándose en los hallazgos clínicos y en las determinaciones hormonales (inicialmente alteración de la GH y el eje tiroideo [TSH] seguido del eje suprarrenal [ACTH] o el eje gonadal [LH/FSH] y, en último lugar, la prolactina). La confirmación del diagnóstico es mediante estudios moleculares o estudio de RMN que tiene un variable espectro lesional (silla turca pequeña, hipófisis hipoplásica o aplásica, ausencia del tallo hipofisario, y ausencia de hiperseñal en la región de la neurohipófisis). El tratamiento del hipopituitarismo será el de los déficits hormonales alterados6.
En resumen, el diagnóstico precoz de este síndrome evita las secuelas neurológicas y permite un desarrollo neuropsicológico adecuado en la edad adulta, por ello, es muy importante tenerlo presente ante un cuadro de retraso psicomotor sin filiar. Sería interesante valorar la realización de la T4 libre (además de la TSH) en el cribado neonatal, ya que podría permitir la detección de hipotiroidismos centrales de forma precoz.
FinanciaciónNo existen fuentes de financiación.

