



El síndrome de Haberland, también conocido como lipomatosis encefalocraneocutánea, es una enfermedad muy infrecuente caracterizada por la existencia de malformaciones neurocutáneas de predominio unilateral que ya se ponen de manifiesto durante las etapas intrauterinas1. Haberland y Perú descubrieron por primera vez esta entidad patológica en el año 1970 en hallazgos clínicos y de necropsia de un hombre de 51 años con epilepsia y retraso mental2. La mayoría de estas lesiones tienen una localización hemicraneal y se asocian generalmente a otras anomalías, como alopecia, quistes aracnoideos, quistes porencefálicos, calcificaciones intracraneales, lipomas intracraneales y de la médula espinal cervical, atrofia cerebral, granulomatosis leptomeníngea, hibrocefalia, polimicrogiria, defectos de laminación cerebral, tumores desmoides en cara y cuero cabelludo, así como anomalías periorbitarias3. La lipomatosis cardiaca también se ha descrito en pacientes con esta enfermedad4. Entre las características clínicas más frecuentes que la suelen acompañar están las crisis epilépticas parciales, la hemiplejía y el retraso mental no progresivo, todas ellas en grados muy variables dependiendo de cada caso en particular. Las crisis epilépticas suelen mostrar un amplio rango de patrones clínicos, estando presentes desde la infancia y afectando a pacientes con grados variables de retraso psicomotor y disfunción motora. En algunas ocasiones los cuadros epilépticos no forman parte de este síndrome, tratándose de casos en los que tampoco existe retraso mental. Otras manifestaciones neurológicas descritas incluyen la espasticidad, la hemiplejía y la paresia facial3,5. Incluso el desarrollo de hemorragias subaracnoideas debidas a malformaciones vasculares han sido referidas por algunos autores6,7. Esta amplia variedad de manifestaciones neurológicas puede dificultar el diagnóstico teniendo en cuenta solo la clínica de los pacientes.
Un paciente varón de 15 años es remitido a nuestro hospital por presentar crisis epilépticas repetidas desde su infancia, alopecia y lesiones de tipo facomatoso en la cara y el cuero cabelludo. Nació en 2001 y su madre presentó un episodio de tipo catarral o seudogripal de relativa entidad durante las etapas finales de gestación, cuya etiología no llegó a ser confirmada. En su historia clínica se refiere también que el parto transcurrió con algunas complicaciones, incluyendo probable distrés fetal durante el mismo. A lo largo de la infancia nuestro paciente presentó numerosas crisis epilépticas, la mayoría de tipo tónico-clónico generalizado y afectando esencialmente al lado derecho de su cuerpo. Casi todas ellas estaban precedidas por un cuadro inflamatorio nasofaríngeo y fiebre, por lo que en un primer momento fueron catalogadas como convulsiones de tipo febril. Sin embargo, en otras ocasiones, los informes neurológicos hacen referencia a crisis focales motoras izquierdas con desviación de la cabeza hacia el mismo lado y en algunas ocasiones se recogen episodios compatibles con cuadros epilépticos de tipo atónico caracterizados por hipotonicidad muscular generalizada, somnolencia y relajación de esfínteres.
Otras alteraciones neurológicas que han persistido hasta el momento actual son la parálisis cerebral, la macrocefalia leve, el retraso del desarrollo psicomotor, el piramidalismo izquierdo y la marcha atáxica central agravada por escoliosis lumbar y dismetría de miembros inferiores.
Dentro de las alteraciones cutáneas destaca la alopecia frontal bilateral y temporal del lado izquierdo, la implantación baja del cabello, así como numerosos nódulos cutáneos polipoideos diagnosticados de fibrolipomas y distribuidos de manera irregular en la cara y el cuero cabelludo.
También consideramos oportuno destacar que nuestro paciente presenta una clinodactilia bilateral que afecta al quinto dedo de ambas manos, retraso de la edad ósea, estrabismo en el ojo derecho y episodios recurrentes de faringoamigdalitis prácticamente desde el nacimiento hasta el día de hoy.
Durante sus primeros años de vida se le hicieron numerosos estudios de imagen, entre los que destacan una TC craneal al año de edad en la que ya se evidenciaba la atrofia cortical cerebral afectando primordialmente a los lóbulos frontales y otra TC craneal a los 5 años en la que se observó la presencia de calcificaciones corticales bilaterales adyacentes a la porción superior y medial de los lóbulos parietales, aparte de la atrofia encefálica ya conocida.
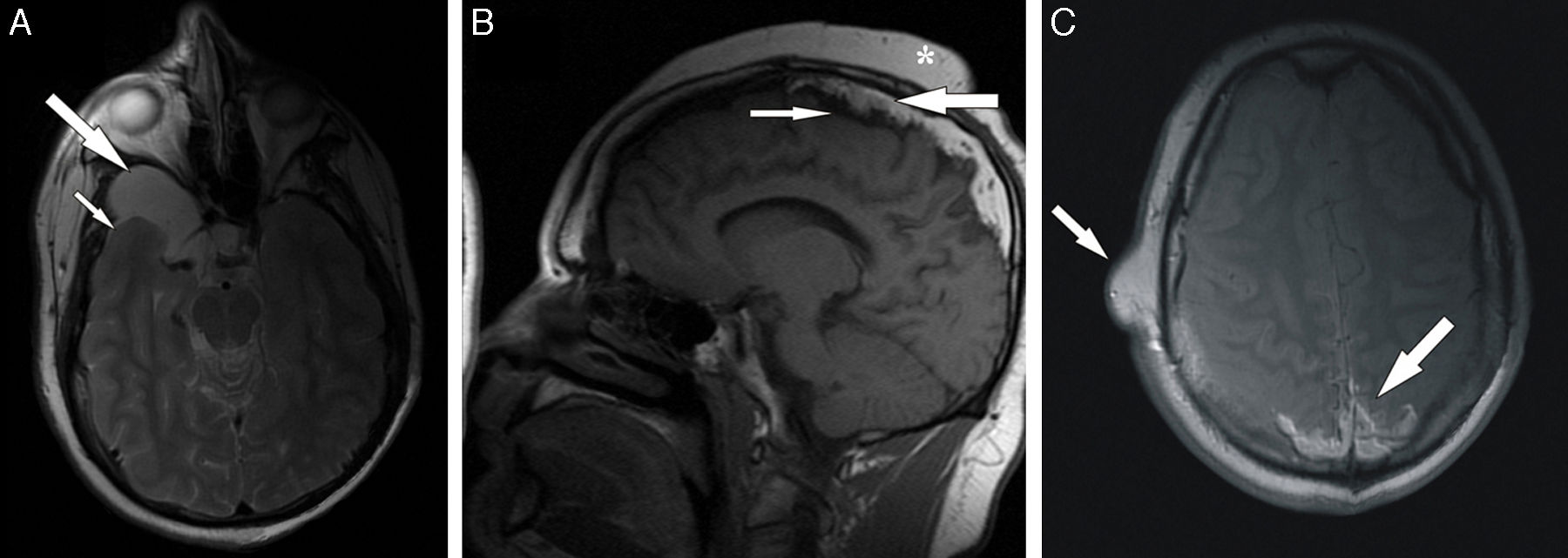
Entre los 10 y los 15 años, los ingresos hospitalarios del paciente disminuyeron drásticamente coincidiendo con la dispensación de tratamiento antiepiléptico asociando fenobarbital y carbamazepina. Tras este periodo, las crisis epilépticas tónico-clónicas aumentaron de nuevo en frecuencia y severidad, lo que condujo a realizar numerosos estudios electroencefalográficos en los que no se encontraron hallazgos específicos de ninguna patología en concreto. Finalmente, coincidiendo con un nuevo empeoramiento del paciente en relación con los episodios convulsivos, este fue remitido al Servicio de Radiodiagnóstico de nuestro hospital para llevar a cabo una resonancia magnética encefálica. Dicha prueba puso de manifiesto numerosos hallazgos que en su conjunto nos condujeron a establecer el diagnóstico definitivo. Por una parte, se aprecia la atrofia cerebral ya conocida y se confirma que la imagen líquida situada en la porción derecha de la fosa craneal media se corresponde con un quiste aracnoideo. Esta última lesión se presenta como un área homogénea, bien delimitada, hipointensa en las secuencias pT1 y moderadamente hiperintensa en las secuencias pT2 (fig. 1). Por otro lado, las calcificaciones cerebrales no son aquí tan evidentes como en la TC, apreciándose como áreas hipodensas parcheadas tanto en secuencias pT1 como pT2, situadas sobre todo en la línea media, bajo la convexidad superior de la calota y afectando a la cortical de ambos lóbulos parietales (fig. 1). Aparte de todo esto, el hallazgo más importante que nos lleva a realizar el diagnóstico final es la acumulación de grasa extraaxial, también localizada esencialmente en la línea media, bajo la convexidad craneal superior, junto a las calcificaciones antes mencionadas y sobre la corteza de los lóbulos parietales y occipitales (fig. 1). Dicha grasa muestra alta intensidad de señal tanto en pT1 como en pT2 y también parece conformar el componente fundamental de las lesiones cutáneas pedunculadas de la cara y del cuero cabelludo.
 Imagen axial craneal de RM pT2 con supresión de la señal del líquido cefalorraquídeo en la que se evidencia atrofia en el lóbulo temporal derecho (flecha pequeña) y una lesión quística hiperintensa bien delimitada situada en la fosa media que corresponde con un quiste aracnoideo (flecha grande). B) Imagen sagital craneal de RM pT1 con hallazgos muy indicativos de síndrome de Haberland. Extensa área hiperintensa localizada adyacente a la convexidad craneal superior y en línea media interhemisférica que representa la lipomatosis intracraneal (flecha grande). Calcificaciones corticales con baja intensidad de señal situada justo bajo la zona de lipomatosis (flechas pequeñas). Tumoraciones subcutáneas diversas en el cuero cabelludo (asterisco). C) Imagen axial craneal de RM pT1 a la altura de los centros semiovales donde el material lipomatoso es claramente visible en la región posterior de la línea media (flecha grande). También se aprecia un tumor benigno de partes blandas sobre el lado derecho de la escama del temporal (flecha pequeña).")
Secciones de RM del caso. A) Imagen axial craneal de RM pT2 con supresión de la señal del líquido cefalorraquídeo en la que se evidencia atrofia en el lóbulo temporal derecho (flecha pequeña) y una lesión quística hiperintensa bien delimitada situada en la fosa media que corresponde con un quiste aracnoideo (flecha grande). B) Imagen sagital craneal de RM pT1 con hallazgos muy indicativos de síndrome de Haberland. Extensa área hiperintensa localizada adyacente a la convexidad craneal superior y en línea media interhemisférica que representa la lipomatosis intracraneal (flecha grande). Calcificaciones corticales con baja intensidad de señal situada justo bajo la zona de lipomatosis (flechas pequeñas). Tumoraciones subcutáneas diversas en el cuero cabelludo (asterisco). C) Imagen axial craneal de RM pT1 a la altura de los centros semiovales donde el material lipomatoso es claramente visible en la región posterior de la línea media (flecha grande). También se aprecia un tumor benigno de partes blandas sobre el lado derecho de la escama del temporal (flecha pequeña).
Como conclusión fundamental destacamos que el síndrome de Haberland, a pesar de ser una enfermedad muy poco común, tiene unas características clínicas y radiológicas que dentro de su variabilidad son bastante típicas. De ahí que tanto los neurólogos como los radiólogos deban hacer una valoración global e integrada de las mismas para establecer su diagnóstico de forma temprana y certera.

