



La epilepsia ausencia infantil (EAI) se considera una forma de epilepsia de fácil control farmacológico solo si se emplean criterios estrictos para la clasificación de los pacientes. Supone el 10% de las epilepsias infantiles de inicio antes de los 15 años y es más frecuente en niñas escolares. El objetivo es conocer la evolución a largo plazo de los pacientes atendidos en la etapa infantil con EAI empleando los criterios de Loiseau y Panayiotopoulos
MétodosEstudio retrospectivo de 69 pacientes con EAI con edad actual mayor de 11 años, realizado mediante revisión de historias clínicas, EEG y cuestionario telefónico.
ResultadosCumplieron los criterios de Loiseau y Panayiotopoulos 52 pacientes, edad actual media 17,61 años. Relación mujeres/hombres: 1,65/1; edad de inicio media: 6 años y 2 meses; duración total de tratamiento media: 3 años y 9 meses; antecedentes familiares de epilepsia: 30,8%; antecedentes personales de crisis febriles: 7,7%; tipo de ausencias: simples 73,5·%, complejas: 26,5%; respuesta al primer tratamiento: ácido valproico 46,3% o ácido valproico con etosuximida simultáneos 90,9%; respuesta al segundo tratamiento (etosuximida o lamotrigina) 84,2%; crisis tras supresión de tratamiento: 4%; pacientes en remisión terminal: 78,8%; necesidad de apoyo psicopedagógico: 25%.
ConclusionesNuestros datos muestran la utilidad de clasificar a los pacientes utilizando criterios estrictos ya que el pronóstico de las crisis del síndrome de EAI puro es excelente. Encontramos que la tasa de recaídas ha sido muy baja. A pesar del favorable pronóstico en cuanto al control de crisis necesitan apoyos psicopedagógicos en un alto porcentaje.
Childhood absence epilepsy (CAE) is considered easily manageable with medication provided that a strict patient classification system is employed. It accounts for 10% of all childhood epilepsy cases starting before the age of 15 and it is most frequent in school-aged girls. The aim of this study is to analyse long-term outcomes of patients diagnosed with CAE according to the Loiseau and Panayiotopoulos criteria and treated during childhood.
MethodsWe conducted a retrospective study including 69 patients with CAE who are currently older than 11; data were gathered from medical histories, EEG records, and telephone questionnaires.
Results52 patients met the Loiseau and Panayiotopoulos criteria. Mean age is now 17.16 years. Female-to-male ratio was 1.65:1; mean age at onset was 6 years and 2 months; mean duration of treatment was 3 years and 9 months. A family history of epilepsy was present in 30.8% of the patients and 7.7% had a personal history of febrile convulsions. Absence seizures were simple in 73.5% of the patients and complex in 26.5%. Response rates to first-line treatment were as follows: valproic acid, 46.3%; and valproic acid plus ethosuximide, 90.9%. The rate of response to second-line therapy (ethosuximide or lamotrigine) was 84.2%; 4% of the patients experienced further seizures after treatment discontinuation, 78.8% achieved seizure remission, and 25% needed psychological and academic support.
ConclusionsOur data show that epileptic patients should be classified according to strict diagnostic criteria since patients with true CAE have an excellent prognosis. The relapse rate was very low in our sample. Despite the favourable prognosis, psychological and academic support is usually necessary.
Las ausencias son un tipo de crisis epilépticas definidas por Willey et al.1 como: Crisis de comienzo brusco que provoca la interrupción de la conciencia. Durante la crisis el paciente queda con la mirada perdida, a veces con elevación de los ojos y parpadeo; si el paciente está hablando, el lenguaje se lentifica o se interrumpe; si está andando se para o deambula torpemente; si come, detiene su mano en el camino hacia la boca y no responde cuando se le habla. A veces, un estímulo sensorial puede abortar la crisis. El episodio termina en unos segundos de forma brusca, tal como había comenzado1. En función de sus características electroclínicas las ausencias se pueden clasificar en típicas y atípicas. En las ausencias típicas se produce una supresión de las funciones mentales, incluyendo comprensión, reactividad y memoria, se caracterizan porque comienzan y terminan de forma brusca y suelen durar entre 5-15seg. En el EEG ictal se recogen descargas generalizadas de punta-onda a 3Hz, sincrónicas y simétricas2. Las ausencias típicas forman parte de numerosos síndromes epilépticos dentro de la Clasificación de la Epilepsia y Síndromes Epilépticos, como la epilepsia ausencia infantil (EAI), el objeto de nuestro estudio, clasificado dentro de las formas generalizadas idiopáticas3.
La EAI se caracteriza por presentar ausencias típicas, tanto simples como complejas, como único tipo de crisis al inicio del cuadro, múltiples a lo largo del día, e inicio antes de la pubertad con desarrollo psicomotor normal y alteraciones en el EEG ictal que se corresponden a descargas de punta-onda regular, bilateral, simétrica y síncrona a 3Hz con actividad de fondo normal o levemente alterada. Es una epilepsia generalizada frecuente en la etapa escolar de 6 a 10 años4, supone entre un 10-17% del total de las epilepsias de diagnóstico en los escolares5, con una prevalencia del 1,5-12% en función de las series6 y, salvo excepciones, es más frecuente en el sexo femenino. En cuanto al pronóstico se han publicado tasas de remisión variable en función de los criterios diagnósticos utilizados. Según los últimos criterios de Panayiotopoulos7 revisados por Loiseau8, que son más restrictivos que los que planteaba la clasificación de la International League Against Epilepsy de 1989, la tasa de remisión puede llegar al 90%. Utilizar estos criterios permite una definición de un grupo más homogéneo de pacientes y hace que podamos conocer mejor la evolución de la EAI9. Clásicamente se ha considerado este síndrome epiléptico dentro de las epilepsias «benignas» por su elevada tasa de control de crisis, pero actualmente sabemos que hasta el 38% de los pacientes con EAI presentarán problemas psicosociales, académicos o laborales10 y que hasta un 35% pueden tener dificultades significativas de atención a pesar de un cociente intelectual normal11.
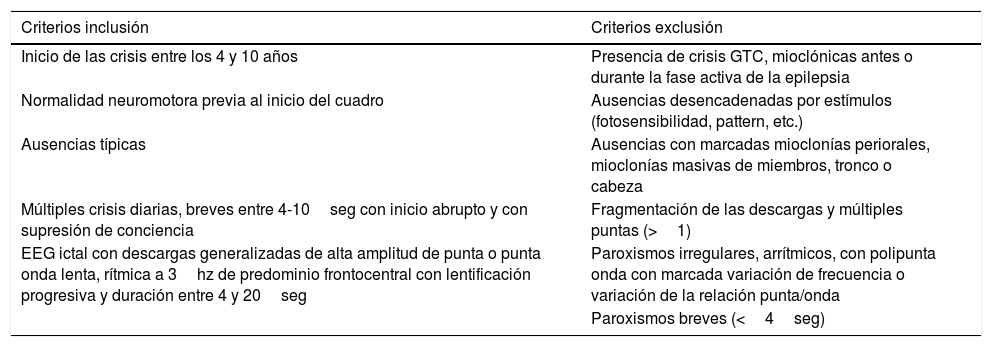
Nuestro objetivo era conocer la evolución a largo plazo de los pacientes atendidos en la etapa infantil con EAI empleando para su selección los criterios de Loiseau y Panayiotopoulos (tabla 1).
Criterios de la International League Against Epilepsy de 1989 y Loiseau y Panayiotopoulos 2005 (adaptados para esta revisión)
| Criterios inclusión | Criterios exclusión |
|---|---|
| Inicio de las crisis entre los 4 y 10 años | Presencia de crisis GTC, mioclónicas antes o durante la fase activa de la epilepsia |
| Normalidad neuromotora previa al inicio del cuadro | Ausencias desencadenadas por estímulos (fotosensibilidad, pattern, etc.) |
| Ausencias típicas | Ausencias con marcadas mioclonías periorales, mioclonías masivas de miembros, tronco o cabeza |
| Múltiples crisis diarias, breves entre 4-10seg con inicio abrupto y con supresión de conciencia | Fragmentación de las descargas y múltiples puntas (>1) |
| EEG ictal con descargas generalizadas de alta amplitud de punta o punta onda lenta, rítmica a 3hz de predominio frontocentral con lentificación progresiva y duración entre 4 y 20seg | Paroxismos irregulares, arrítmicos, con polipunta onda con marcada variación de frecuencia o variación de la relación punta/onda |
| Paroxismos breves (<4seg) |
EEG: electroencefalograma; crisis GTC: crisis generalizadas tónico-clónicas.
Diseñamos un estudio retrospectivo sobre los pacientes que fueron diagnosticados de EAI entre los años 1988 y 2015 que tienen una edad actual mayor de 11 años. Los pacientes provienen de 2 consultas de Neuropediatría, una hospitalaria y otra extrahospitalaria, que atienden pacientes con características demográficas y clínicas similares, donde los criterios de diagnóstico y de tratamiento son homogéneos. En el estudio se han recogido de la historia clínica los siguientes datos: sexo, historia familiar de epilepsia, crisis febriles del paciente, tipo de ausencias, edad de inicio de las ausencias, tiempo de evolución hasta el diagnóstico, fármacos utilizados y respuesta a los mismos, EEG al diagnóstico, evolución del mismo y duración del tratamiento. Por otra parte, realizamos un cuestionario telefónico voluntario, estructurado, sobre la existencia de recidiva de crisis tras retirar el tratamiento, tipo de las mismas, remisión terminal (para nuestro estudio, un año sin tratamiento y sin crisis), tratamiento actual, necesidad de apoyo psicopedagógico y problemas psicológicos.
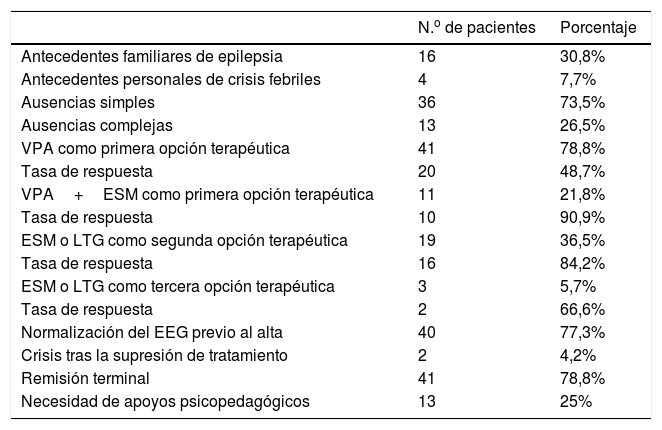
ResultadosSe identificaron 69 pacientes diagnosticados de EAI entre los años 1988 y 2015 cuya edad, en el momento del realizar el estudio, era mayor de 11 años. Aplicando los criterios de Loiseau y Panayiotopoulos, fueron excluidos 17, por lo que, finalmente, el número de pacientes objeto del estudio fue de 52. Al cuestionario telefónico sobre la evolución a largo plazo respondieron 47 (90%); no pudieron ser localizados 5 pacientes. La edad media del grupo fue de 17,61 años (DE 5,79) con una edad mínima de 11 y máxima de 36 años; la edad media del inicio de las ausencias fue de 6 años (73,8 meses; DE 21,77) con una edad mínima de 4 años y máxima de 9 años y 6 meses. La distribución por sexos fue de 32 mujeres (62%) y 20 hombres (38%) con una relación 1,65/1.
Tenían antecedentes familiares de epilepsia 16 pacientes (30,8%) y antecedentes personales de crisis febriles 4 (7,7%). En cuanto al tipo de ausencias, tenían ausencias simples 36 (73,5%) y ausencias complejas 13 pacientes (26,5%). El tiempo de evolución desde el inicio del cuadro hasta la primera consulta fue de 6 meses (25,63 semanas; DE 24,12) y la duración media de tratamiento de 3 años y 9 meses (45,31 meses; DE 31,01).
El tratamiento inicial fue ácido valproico (VPA) en monoterapia en 41 pacientes de los que el 48% quedaron libres de crisis. En un grupo de 11 niños se pautó, desde el inicio, la asociación de VPA con etosuximida (ESM) con una tasa de respuesta del 90,9%. Tras la primera línea de tratamiento, 19 pacientes continuaron con crisis, en estos el segundo fármaco usado fue ESM en 16 y lamotrigina (LTG) en 3, consiguiendo el control de crisis en 16 (tasa de respuesta del 84,2%). Los 3 pacientes restantes necesitaron un tercer fármaco (ESM o LTG) al que respondieron 2 de ellos (66%). La duración media del tratamiento fue 3,5 años (máximo 7 años). Durante el seguimiento, persistieron las anomalías en el EEG en un 22,7%. Solo 2 pacientes tuvieron crisis tras la supresión del tratamiento (4%), en un caso volvió a tener crisis de ausencias y en otro mioclónicas. La remisión terminal se consiguió en el 78,8%. Necesitaron apoyo psicopedagógico el 25% de los pacientes (tabla 2).
Resultados
| N.o de pacientes | Porcentaje | |
|---|---|---|
| Antecedentes familiares de epilepsia | 16 | 30,8% |
| Antecedentes personales de crisis febriles | 4 | 7,7% |
| Ausencias simples | 36 | 73,5% |
| Ausencias complejas | 13 | 26,5% |
| VPA como primera opción terapéutica | 41 | 78,8% |
| Tasa de respuesta | 20 | 48,7% |
| VPA+ESM como primera opción terapéutica | 11 | 21,8% |
| Tasa de respuesta | 10 | 90,9% |
| ESM o LTG como segunda opción terapéutica | 19 | 36,5% |
| Tasa de respuesta | 16 | 84,2% |
| ESM o LTG como tercera opción terapéutica | 3 | 5,7% |
| Tasa de respuesta | 2 | 66,6% |
| Normalización del EEG previo al alta | 40 | 77,3% |
| Crisis tras la supresión de tratamiento | 2 | 4,2% |
| Remisión terminal | 41 | 78,8% |
| Necesidad de apoyos psicopedagógicos | 13 | 25% |
VPA: ácido valproico; ESM: etosuximida; LTG: lamotrigina; EEG: electroencefalograma.
Existen múltiples trabajos que analizan la evolución a largo plazo de la EAI, pero emplean distintos criterios de clasificación y metodología, por lo que los datos hallados en la literatura son variables. Ante esta evidencia, decidimos utilizar en nuestro trabajo unos criterios estrictos7,8, en concreto los de Loiseau y Panayiotopoulos, para seleccionar una muestra homogénea a pesar de la procedencia de 2 centros diferentes. Presentamos una serie de EAI «pura» de mayor tamaño que las publicadas en nuestro medio4,12. El carácter retrospectivo del estudio conlleva las limitaciones propias de este tipo de estudios: sesgos de recogida de datos y sesgos de memoria que pueden afectar a los resultados.
La EAI comienza, por lo general, entre los 4 y 10 años, con un pico entre 5-7 años13 y, salvo excepciones, es claramente más frecuente en el sexo femenino14. En nuestra serie encontramos también estas características, los pacientes tenían una media edad de 73,8 meses (6 años), predominio del sexo femenino (62% de niñas), lo que caracteriza a la EAI como una epilepsia propia de niñas en edad escolar. La edad media de 17,61 años en el momento del estudio es la consecuencia de la revisión a largo plazo de la evaluación realizada. La frecuencia de antecedentes familiares de epilepsia (30,8%) y de antecedentes de crisis febriles (8%) concuerda con las ya publicadas, reflejando una base genética predisponente.
En nuestro estudio, el 73,5% tenían ausencias simples y el 26,5% ausencias complejas, similar a la bibliografía consultada. El tiempo de evolución desde el inicio del cuadro hasta la primera consulta fue de más de 6 meses (25,63 semanas), probablemente debido al carácter sutil de las crisis que a menudo las hace pasar desapercibidas o se diagnostican erróneamente como tics, despistes o movimientos estereotipados.
Como sabemos, los fármacos más usados para EAI han sido VPA, ESM y LTG pero hasta 2010 solo se habían realizado 6 ensayos, con escaso número de pacientes, para examinar la eficacia de los mismos en monoterapia inicial y, dadas las limitaciones metodológicas, ninguno tenía evidencia suficiente para ofrecer recomendaciones sólidas en la práctica clínica15. En 2010 se inició un estudio multicéntrico con 453 pacientes, aleatorizado, controlado y doble ciego comparando la eficacia, tolerabilidad y efectos secundarios de VPA, ESM y LTG. Este estudio evidenció que VPA no tiene una mayor eficacia que ESM o LTG y presenta mayor grado de afectación cognitiva que ESM16. Algunos estudios en ratas, con 2 modelos genéticos diferentes de EAI, muestran que ESM no solo controla las crisis sino que podría tener un «papel modificador de la enfermedad»17–18. En este sentido, en 2014 se realizó un estudio prospectivo para valorar la evolución a largo plazo de los pacientes según el tratamiento inicial utilizado (VPA o ESM) y se evidenció una clara tendencia a mantener mayor tasa de remisión completa si los pacientes se tratan con ESM en lugar de VPA, independientemente de las atipicidades de EEG19. En nuestra serie empleamos VPA como primer fármaco, solo o asociado a ESM, en todos los pacientes. La eficacia de VPA ha sido modesta comparada con los datos encontrados en la literatura (el 48,7% en nuestra serie comparada con el 77-85% de la literatura)12,20, posiblemente porque recurrimos al uso de terapia combinada VPA con ESM si no había respuesta inicial, sin aumentar VPA a las dosis comunicadas en otros trabajos. Además, nos parece interesante dar a conocer que en los pacientes en los que se inició el tratamiento con terapia combinada (VPA+ESM) se logró el control del 90,9% en el primer intento, sin aumento de efectos adversos. Revisando la literatura no hemos hallado estudios similares por lo que no podemos comparar estos resultados.
No existen criterios unánimes sobre la duración del tratamiento y, en general, se recomienda entre uno y dos años sin crisis y con EEG normalizado como criterio para iniciar la supresión15. Hemos comprobado que en nuestros pacientes la media de duración es superior (3,5 años), con casos de hasta 7 años, lo que indica que, a pesar del buen pronóstico de EAI, es necesaria una larga vigilancia especialmente en los casos con mala respuesta inicial.
Las recaídas con crisis de ausencia tras la supresión de tratamiento ocurren en el 8% cuando se analizan a pacientes con EAI pura9. Es posible que nuestra cifra del 4% esté influida por el carácter retrospectivo del estudio o por la mayor duración del tratamiento.
El porcentaje de pacientes que desarrollan crisis tonicoclónicas generalizadas en la etapa juvenil se ha establecido entre el 35-60% según los diferentes estudios21 y suelen ocurrir unos 5-10 años tras el inicio de las ausencias22. Nosotros no hemos tenido ningún paciente con crisis tonicoclónicas generalizadas antes de la remisión terminal.
En cuanto a la tasa de remisión terminal en este tipo de epilepsia, es alta, pero varía en función de la metodología empleada y el concepto de remisión terminal utilizado en cada estudio, se han publicado unas tasas entre 50-90%. Considerando la remisión terminal como permanecer un año sin crisis y sin tratamiento, y comparada con trabajos retrospectivos similares al nuestro, el 78,8% de los pacientes con remisión terminal es coincidente con la cifra publicada en el 2005 por Grosso et al.9.
Se ha insistido siempre en el buen comportamiento evolutivo de la EAI, sin embargo hemos podido comprobar, al igual que otros autores10,11, que el porcentaje de pacientes con dificultades psicopedagógicas es muy elevado, en nuestro caso del 25%. Según otras series hasta el 38% de los pacientes con EAI presentarán problemas psicosociales, académicos o laborales10. Y hasta un 35% de los pacientes pueden tener dificultades significativas de atención a pesar de un cociente intelectual normal11. Estos problemas de atención interfieren en la memoria y en las funciones ejecutivas y, por ende, en el rendimiento académico, pero en la mayoría de los casos esta falta de atención no es detectada por los padres. Además, en estudios realizados con pocos sujetos, se han detectado dificultades visoespaciales, del aprendizaje verbal y la memoria23,24 y de las habilidades del lenguaje25.
Podemos concluir que el diagnóstico de EAI con criterios estrictos, como los de Loiseau y Panayiotopoulos, permite ofrecer un pronóstico favorable. En nuestra serie, el tratamiento se ha prolongado en algunos pacientes más de lo aconsejado en las recomendaciones clásicas, posiblemente para prevenir recaídas. Consideramos que la ESM puede ofrecerse como primer fármaco en el tratamiento de EAI diagnosticada con criterios estrictos. Destacamos la presencia de un notable porcentaje de pacientes con dificultades psicopedagógicas que persisten a pesar del control de las crisis y de la supresión de la medicación antiepiléptica.
Conflicto de interesesNo hay conflicto de intereses.

