



El ictus es una de las principales causas de mortalidad en el mundo y debido al incremento en la expectativa de vida su incidencia va en aumento; sin embargo, el desarrollo de nuevos medicamentos con utilidad clínica ha sido prácticamente nulo, por lo que hasta la fecha el tratamiento de estos pacientes es muy limitado.
DesarrolloLa evidencia básica y clínica en el área señala que tras un infarto cerebral se producen una serie de cambios neuroquímicos, entre los que se encuentran: la depleción energética, la producción de radicales libres, la acumulación de calcio, la desregulación de neurotransmisores, la excitotoxicidad, y de manera tardía, la activación del sistema inmune caracterizada como inflamación. Esta respuesta del sistema inmunológico ha mostrado ser un evento central en la progresión de la patología, en el que destaca la participación de las citocinas proinflamatorias como TNF, que aumentan el daño por excitotoxicidad y por acumulación de calcio, favorecen la formación de radicales libres y en general promueven la muerte celular. Por otro lado, algunas citocinas antiinflamatorias como IL-10 e IL-4 han mostrado tener efectos neuroprotectores e incluso favorecen la recuperación de sinapsis y la neurogénesis, haciendo de la modulación de la respuesta inmunológica un área con mucho potencial terapéutico.
ConclusionesEl entendimiento de las relaciones entre el sistema inmunológico y el sistema nervioso no solo nos permite entender con mayor profundidad el fenómeno del ictus, sino que también nos ofrece un nuevo arsenal de estrategias diagnósticas, pronósticas y terapéuticas que podrían mejorar la calidad de vida de las personas aquejadas por esta terrible enfermedad.
Stroke is one of the leading causes of death in the world; its incidence is increasing due to increased life expectancy. However, treatment options for these patients are limited since no clinically effective drugs have been developed to date.
DevelopmentAccording to clinical evidence, a number of neurochemical changes take place after stroke, including energy depletion, increased free radical synthesis, calcium accumulation, neurotransmitter imbalance, excitotoxicity, and, at a later stage, immune system activation leading to inflammation.
Immune response has been shown to be a major factor in disease progression. The release of proinflammatory cytokines such as TNF increase brain damage secondary to excitotoxicity and calcium accumulation, and promote free radical synthesis and cell death through various mechanisms. On the other hand, certain anti-inflammatory cytokines, such as IL-10 and IL-4, have been shown to have a neuroprotective effect and even promote neurogenesis and synapse remodeling, which makes immune modulation a promising treatment approach.
ConclusionsUnderstanding the relationship between the immune system and the nervous system not only deepens our knowledge of stroke but also provides new diagnostic, prognostic, and therapeutic strategies that may increase the quality of life of stroke patients.
El ictus es la segunda causa de muerte y tercera causa de discapacidad en el mundo. Se estima que 16 millones de personas sufren un ictus isquémico cada año, de los cuales 6 millones mueren, y la mayoría de los pacientes quedan con secuelas importantes. Además, en muchos países, principalmente en aquellos en vías de desarrollo, esta incidencia va en aumento1. Esta patología se caracteriza por un cese en el flujo sanguíneo en una región del cerebro, en la mayoría de los casos debido a un coágulo o trombo que obstruye los vasos cerebrales. Esta obstrucción en el flujo sanguíneo genera que las células del área mueran formando un área llamada núcleo isquémico, mientras que el tejido circundante queda en un estado de hipoperfusión, también conocido como penumbra isquémica; esta última es especialmente importante debido a que la muerte progresiva de esta área o su recuperación determina en gran parte el pronóstico y la recuperación funcional del paciente2.
En este artículo revisaremos la importante relación que existe entre la activación del sistema inmunológico y el destino final de la zona de penumbra, así como las alteraciones propias del sistema inmune en respuesta al ictus.
Fisiopatología en el ictus isquémicoCon respecto a este tema existen publicadas extensas revisiones de la fisiopatología del ictus; entre estas sugerimos revisar las referencias 3, 5, 6, 7, y 8, pues aquí solo mencionaremos brevemente las generalidades.
Una vez que el flujo sanguíneo ha sido interrumpido, la falta de oxígeno y glucosa en el tejido llevan a una rápida depleción del trifosfato de adenosina (ATP), la principal molécula energética en el cuerpo3. Esta disminución en las concentraciones de ATP impide la realización de funciones esenciales para la célula; por ejemplo, la bomba Na+/K+ ATPasa (sodio/potasio ATPasa) pierde su función, generando alteraciones en el potencial de membrana en reposo y acumulación de Na+ intracelular, lo que a su vez induce la despolarización anóxica y el edema citotóxico4. Esta despolarización anóxica se propaga por la zona de penumbra4, generando una liberación no regulada y dañina de neurotransmisores como el glutamato5, que se cree que participa aumentando el daño del tejido cerebral4.
Al continuar la despolarización aumenta, de manera importante, la concentración del neurotransmisor glutamato, que al activar el receptor NMDA provoca un aumento en la conductabilidad al Na+, que por su efecto osmótico causa un edema citotóxico. Además, la hiperactividad del receptor NMDA aumenta también la concentración de Ca++, que puede generar disfunción mitocondrial al aumentar su permeabilidad, incrementar la producción de radicales libres, activar las enzimas fosfolipasas y proteasas llamadas caspasas, propiciando la muerte celular por apoptosis5,6.
De manera un poco más tardía, los componentes celulares de las células dañadas pueden activar las células del sistema inmunológico promoviendo la liberación de citocinas que mencionaremos más adelante, pero promueven la síntesis de radicales libres como el óxido nítrico (NO) con su eventual metabolismo a nitrito (NO2) y al radical hidroxilo (OH•)7; esto, aunado al daño por la isquemia, puede generar una pérdida de la permeabilidad selectiva de la barrera hematoencefálica (BHE), permitiendo la entrada de sustancias potencialmente tóxicas al sistema nervioso central, además de reclutar un mayor número de células del sistema inmunológico a través de moléculas de adhesión (ICAM y VCAM)8,9.
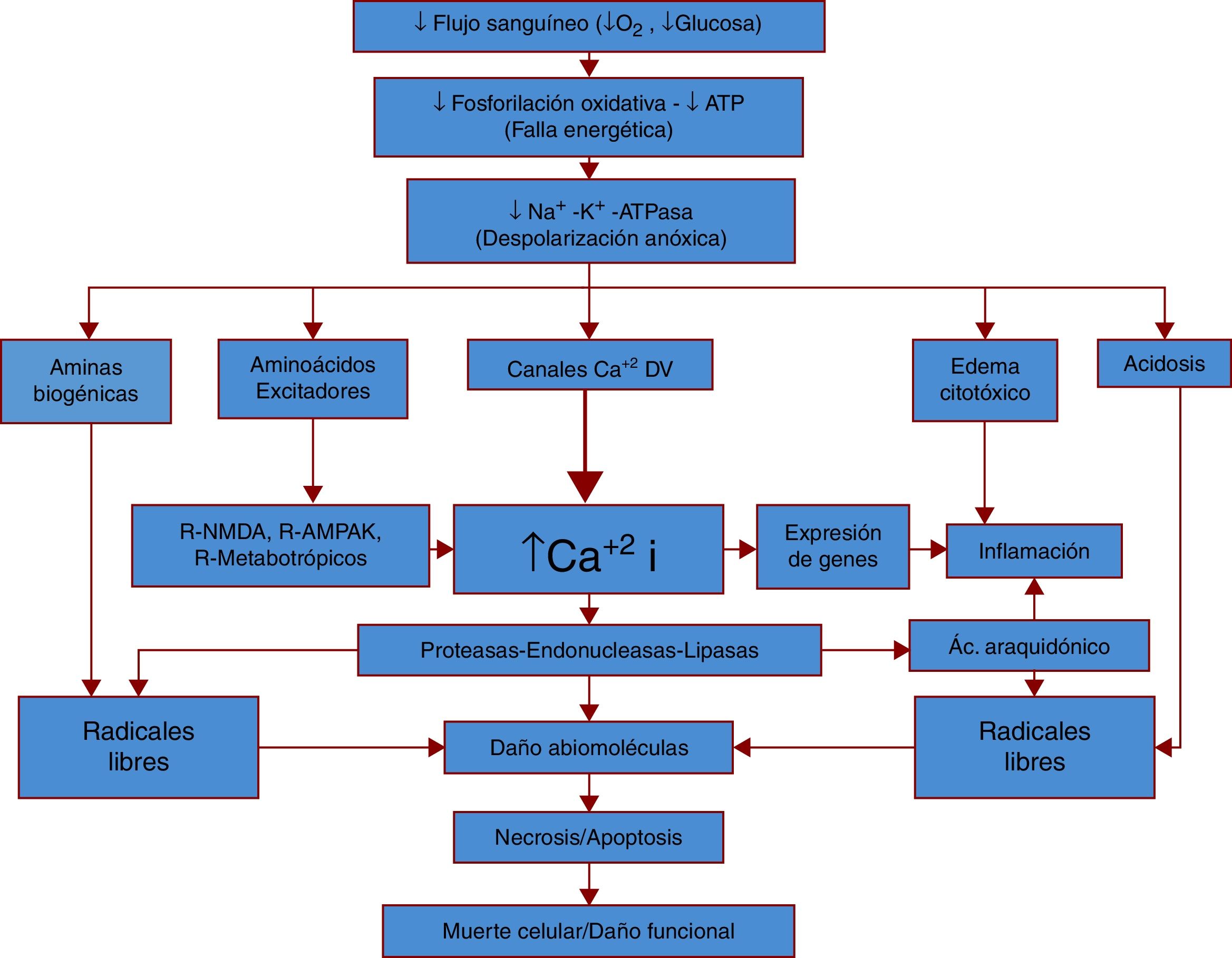
La figura 1 representa de manera simplificada la fisiopatología general del ictus isquémico.
, llevando a una disfunción en numerosos procesos, siendo uno de los más importantes la bomba Na+/K+ ATPasa, la cual al presentar disfunción genera despolarizaciones anóxicas. Al darse estas despolarizaciones sin los elementos metabólicos necesarios para su control, la neurona sufre una acumulación de neurotransmisores y otras aminas, lo que conlleva un aumento tóxico de radicales libres, calcio, agua e hidrogeniones. En respuesta se genera una respuesta inflamatoria importante y estos cambios llevan a la muerte celular y al daño funcional.")
Fisiopatología general del ictus isquémico.
La fisiopatología del ictus comienza con una diminución importante del flujo sanguíneo y por lo tanto de oxígeno y glucosa, sustratos energéticos esenciales para el funcionamiento neuronal. Con la pérdida de estos sustratos disminuye la producción energética (ATP), llevando a una disfunción en numerosos procesos, siendo uno de los más importantes la bomba Na+/K+ ATPasa, la cual al presentar disfunción genera despolarizaciones anóxicas. Al darse estas despolarizaciones sin los elementos metabólicos necesarios para su control, la neurona sufre una acumulación de neurotransmisores y otras aminas, lo que conlleva un aumento tóxico de radicales libres, calcio, agua e hidrogeniones. En respuesta se genera una respuesta inflamatoria importante y estos cambios llevan a la muerte celular y al daño funcional.
El daño celular general y la liberación de distintos componentes celulares darán lugar a una reacción inflamatoria en el sitio del cerebro afectado; los componentes de esta reacción modularán de manera importante la respuesta de la región de penumbra, pudiendo agravar el daño o ser neuroprotectores.
Uno de los primeros pasos de esta respuesta es la detección de patrones moleculares de daño (damage associated molecular patterns [DAMP]) por parte de los receptores del sistema inmunológico innato tipo toll (toll like receptors [TLR]) presentes principalmente en la microglía, aunque también en menor concentración en el endotelio y los astrocitos10,11. La activación de varios de estos receptores lleva a la secreción de numerosos componentes importantes de la respuesta inmunológica, entre los que encontramos citocinas, quimiocinas, la inducción enzimática de la óxido nítrico sintasa (iNOS) y de la ciclo oxigenasa 2 (COX2), con el aumento subsecuente de óxido nítrico y prostaglandina E2 (PGE2), mieloperoxidasas y el reclutamiento de más componentes celulares y no celulares del sistema inmunológico10,12.
La importancia de los TLR ha sido demostrada en ensayos experimentales de obstrucción de la arteria cerebral media, donde un ratón mutante que no expresa TLR 4 presenta infartos más pequeños, así como un mejor estado neurológico en una batería estandarizada, además de menor actividad de iNOS y COX2, gelatinasa B (proteasa implicada en la transformación hemorrágica de los infartos cerebrales) y un nivel menor de malondialdehído, importante marcador de actividad de radicales libres; sin embargo no hubo diferencias significativas en los niveles de citocinas12.
A pesar de la protección que presenta el ratón mutante para TLR 4, su papel no es tan sencillo; por ejemplo, al administrar lipopolisacárido (LPS) —un producto bacteriano que activa al TLR 4— previo al daño isquémico, este resulta neuroprotector en un fenómeno conocido como precondicionamiento13.
Durante el precondicionamiento, la exposición al LPS modifica la respuesta del TLR 4 al reclutar la vía de señalización TRIF-IRF3, en lugar de la MyD88 clásica para este receptor, causando un aumento en la concentración de interferón beta (INF-β), que como veremos más adelante tiene un importante potencial neuroprotector13.
Ahora revisaremos algunos de los componentes específicos de la respuesta inmunológica y su papel en el infarto cerebral.
AstrocitosComo parte integral de la sinapsis los atrocitos responden de manera temprana al evento isquémico. Como hemos mencionado previamente, estas células pueden ser activadas por moléculas asociadas al daño celular a través de los receptores tipo toll, así como también por la presencia de citocinas en el medio. Por cualquiera de los 2 mecanismos, el evento isquémico induce en los astrocitos una respuesta llamada astrogliosis, siendo de especial importancia el segundo mensajero STAT 3 que genera el cambio fenotípico14.
La astrogliosis puede aislar el tejido dañado e impedir la propagación de este daño al generar una banda alrededor del sitio de insulto, captura células proinflamatorias, previene el aumento de volumen del infarto y mejora el pronóstico funcional en animales14,15.
La ablación de STAT 3 asociada al segundo mensajero específico de glía, la proteína ácida glial fibrilar (GFAP), disminuye la migración de estas células así como su capacidad de aislar el tejido dañado en un modelo de daño medular14, mientras que en un modelo donde se inhibía al segundo mensajero Socs3 (principal inhibidor de STAT 3) aumentando su actividad, los ratones presentaban una mejor contracción del tejido dañado y una mayor recuperación funcional16.
Los mecanismos de neuroprotección mediados por el astrocito son diversos. Entre ellos se encuentra la activación de importantes antioxidantes como el glutatión17, la secreción de neurotropinas como GDNF que inhiben el proceso de apoptosis18,19 y mediante el metabolismo de numerosos neurotransmisores, principalmente el glutamato, para prevenir la excitotoxicidad entre otros20,21. Para una revisión detallada de la función y cambios de los astrocitos sugerimos consultar la referencia 20.
Es esta última propiedad de las células astrogliales la que se ha buscado explotar de manera más efectiva; es decir, se ha observado que el aumento dirigido del transportador GLT1 en ratones disminuye el volumen de tejido infartado, mejorando el pronóstico funcional del animal al reducir la concentración de glutamato que se vierte al tejido no infartado y en general disminuyendo la muerte celular después de la ligadura de la arteria cerebral media21.
En este sentido varios medicamentos han mostrado compartir este mecanismo de acción, haciéndolos interesantes dianas terapéuticas a estudiar: el tamoxifeno22, ya usado en la profilaxis de cáncer de mama, el riluzol23, usado en la esclerosis lateral amiotrófica, y el antibiótico ceftriaxona24; este último ha sido usado en modelos de infarto cerebral, mostrando una mejoría en la supervivencia y la recuperación funcional24. A pesar de la función protectora que la astroglía ha demostrado, también puede aumentar el daño, al secretar citocinas proinflamatorias como TNF, IL-6, etc. Además de secretar proteoglicanos condroitin sulfato, un importante inhibidor del crecimiento axonal y la sinaptogénesis, la administración de condroitinasa ha mostrado mejorar la recuperación funcional posterior a un daño espinal25.
Microglía/macrófagosLas células de la microglía son el principal componente del sistema inmunológico presente en el cerebro; de manera basal presentan una movilización extensa por toda la masa encefálica y son capaces de responder de una manera progresiva a diferentes señales de daño (para una revisión detallada ver la referencia 26). Entre los principales determinantes de la función microglial se encuentra el microambiente citocinérgico, donde la activación por INF-γ causa una polarización hacia un fenotipo altamente proinflamatorio y asociado a neurodegeneración (M1), mientras que la IL-4 permite la diferenciación hacia un estado que favorece la neurogénesis, oligodendrogénesis y resulta antiinflamatorio (M2)26,27.
A pesar de la dualidad característica de las células microgliales, su papel durante el infarto cerebral parece ser en general protector; por ejemplo, en un modelo que permite detener a las células microgliales durante su proliferación usando ganciclovir (se usó un ratón mutante que expresa el gen del virus herpes simple acoplado al promotor de células mieloides CD11b), al impedir la proliferación microglial se encontró un aumento del tamaño del infarto, del número de neuronas en apoptosis y de las alteraciones conductuales, lo cual no sucedía en los animales sin la mutación que recibían ganciclovir28.
Al estudiar la dinámica de las células de la microglía en un modelo de oclusión de la arteria cerebral media en ratones, se encontró que la expresión de genes del fenotipo M2 prevalece del día 1 hasta el día 5 postisquemia, mientras que los genes típicos del fenotipo M1 empiezan a expresarse el día 3 y se mantienen hasta el día 1427. Esto adquiere especial relevancia al considerar que en experimentos donde se cultivan neuronas en condiciones de privación de oxígeno y glucosa, y se les agrega microglía que ha sido diferenciada hacia M1, la supervivencia neuronal se reduce de manera importante, mientras que si han sido diferenciadas hacia M2 la supervivencia aumenta27.
Como estrategia terapéutica, el uso de las células microgliales ha encontrado el avance más significativo con el uso del antibiótico minociclina, cuya administración ha demostrado neuroprotección, menor apoptosis neuronal, menor volumen de infarto y mejor recuperación funcional en ratones29.
Debido a la seguridad de este medicamento, así como a la experiencia ya existente en el ámbito de la clínica, la minociclina es una fuente activa de investigación en humanos como tratamiento neuroprotector en el ictus, sin embargo ha arrojado resultados mixtos30,31.
Otros componentes celularesDebido al proceso inflamatorio y a la disfunción de la barrera hematoencefálica, las células del sistema inmunológico periférico pueden acceder al cerebro, modificando el proceso patológico; esto se ha visto que sucede tan pronto como a las 24h, siendo los neutrófilos de los primeros que aparecen32, surgiendo a continuación macrófagos a partir del día 3 y hasta el día 733, y de manera un poco más tardía, otros componentes como los linfocitos T y B. Cada uno de estos grupos celulares ha mostrado una participación muy importante en la fisiopatología del infarto cerebral.
Los neutrófilos, como ya hemos mencionado, son de las primeras células en responder. Estos han sido evidenciados como esencialmente neurotóxicos, y en seres humanos se ha correlacionado la cuenta de neutrófilos en plasma con un peor pronóstico después de un infarto cerebral34, mientras que en modelos animales, el tratamiento con el anticuerpo monoclonal RP3 (el cual es específico para neutrófilos) previene su infiltración al sistema nervioso central, reduce el edema y disminuye el volumen del infarto35,36.
Al ser activados, los neutrófilos generan una disfunción importante y permanente de la BHE; esto es en gran parte debido a la liberación de gelatinasa (metaloproteinasa de la matriz extracelular 9 [MMP 9]), del cual son la principal fuente36,37. La presencia de la gelatinasa y la disfunción de la BHE han mostrado ser partes esenciales en la transformación de un infarto de isquémico a hemorrágico en animales y humanos38,39.
En modelos animales con ratones mutantes que sobreexpresan el inhibidor de metaloproteinasas TIMP-1, tras realizarles una ligadura de arteria carótida presentan una neuroprotección robusta en comparación con los ratones silvestres, con infartos más pequeños, una mayor integridad de la BHE y menor infiltración por leucocitos39,40.
Los linfocitos, a diferencia de las otras células del sistema inmunológico, han sido asociados con una mayor recuperación postinfarto cerebral en la escala NIHSS34. Entre los linfocitos, los más estudiados han sido los T helper (TH); estos, al igual que la microglía, presentan una respuesta polarizada en la que los TH1 secretan principalmente TNF, INF e IL-6, mientras que los TH2 secretan IL-441.
Esta diferenciación funcional no solo afecta la progresión del infarto cerebral y la recuperación funcional (siendo el TH2 neuroprotector), sino que también dirige la respuesta inmunológica periférica a otros insultos. Se ha demostrado que entre las manifestaciones postinfarto cerebral hay un número menor de linfocitos T en el bazo, en el timo, en los nódulos linfáticos y en la barrera hematointestinal, posiblemente debido a un nivel elevado de apoptosis por la actividad del sistema nervioso simpático. La polarización TH2 y la reducción absoluta en el número de linfocitos predispone a infecciones subsecuentes, fenómeno llamado inmunosupresión postinfarto cerebral42-44.
Factor de Necrosis tumoral alfa (TNF-α)El TNF, como hemos visto antes, es de las primeras citocinas secretadas en el infarto cerebral y tiene implicaciones importantes en la supervivencia o muerte celular.
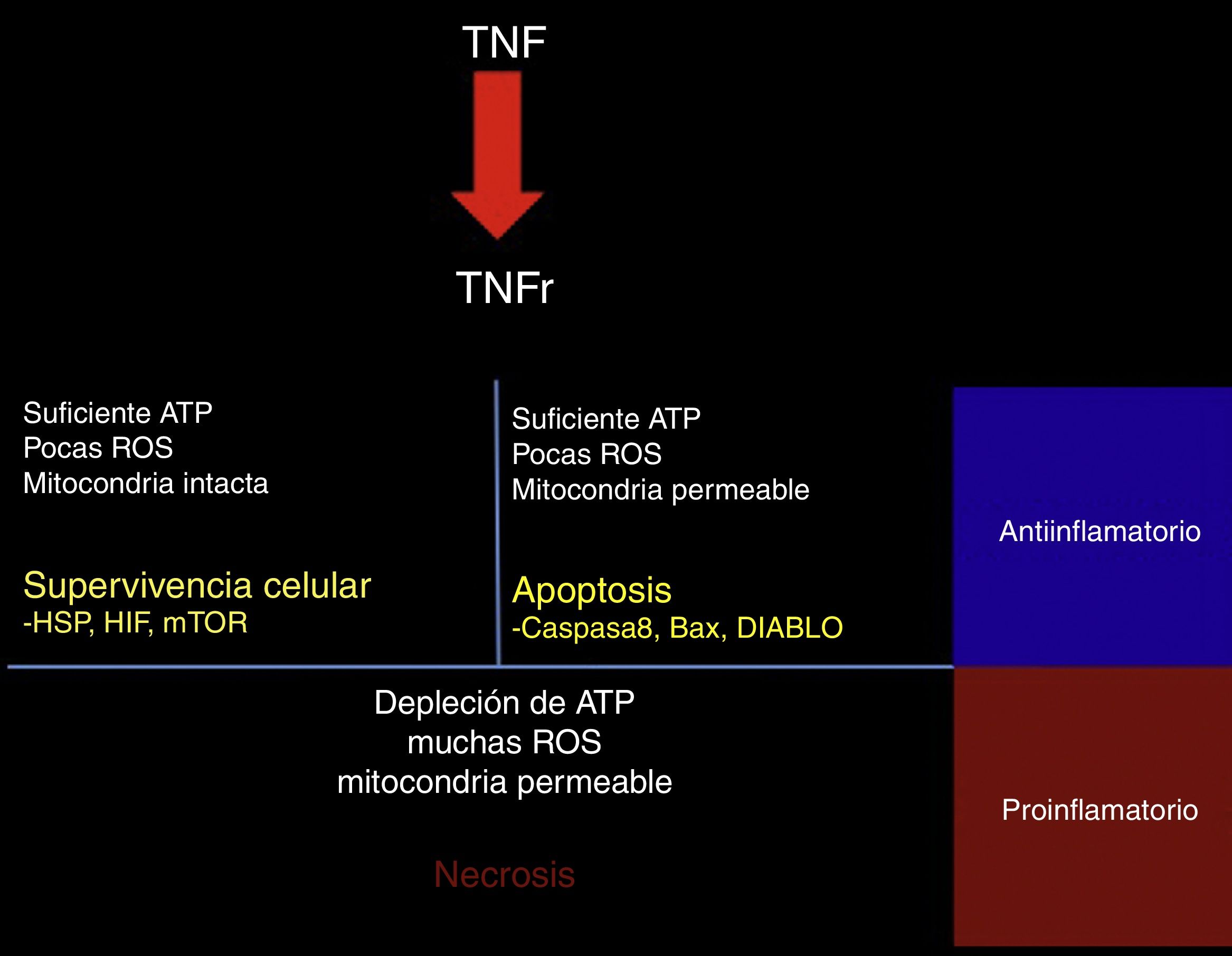
El TNF-α puede tener efectos protectores o citotóxicos dependiendo del medio celular en el que se encuentre; al ser evaluado a través de un modelo matemático, con las diferentes interacciones se encontró que la activación de su receptor TNFR conlleva la activación de una de las 3 vías de señalización. Primero la vía RIP1-NFkB, asociada a supervivencia celular, inhibición de la apoptosis y necrosis, e incluso a un aumento en la resistencia al daño subsecuente. Esta vía se ve favorecida por un medio con baja actividad de especies reactivas al oxígeno (ROS), niveles elevados de ATP y una alta actividad de NFkB45 (fig. 2).
 y la membrana mitocondrial permanece impermeable, la célula activa las proteínas de choque térmico (HSP), el factor inducible de hipoxia (HIF) y el blanco de la rapamicina de mamíferos (mTOR). Si presenta suficiente ATP, pocas ROS, pero la mitocondria es permeable, se activan las vías proapoptóticas a través de los mediadores caspasa 8, Bax y DIABLO. Por último, si hay una profunda depleción de ATP, una gran cantidad de ROS y una mitocondria en franca disfunción, la célula hará necrosis. En el contexto del tejido, las células que sobreviven o que hacen apoptosis generan un estímulo antiinflamatorio y citoprotector, mientras que la necrosis es un importante estímulo proinflamatorio.")
Respuesta al TNF.Al activarse, el receptor de TNF activa mecanismos de señalización intracelular dependientes del estado metabólico de la célula; si presenta aún suficiente ATP, pocas especies reactivas de oxígeno (ROS) y la membrana mitocondrial permanece impermeable, la célula activa las proteínas de choque térmico (HSP), el factor inducible de hipoxia (HIF) y el blanco de la rapamicina de mamíferos (mTOR). Si presenta suficiente ATP, pocas ROS, pero la mitocondria es permeable, se activan las vías proapoptóticas a través de los mediadores caspasa 8, Bax y DIABLO.
Por último, si hay una profunda depleción de ATP, una gran cantidad de ROS y una mitocondria en franca disfunción, la célula hará necrosis. En el contexto del tejido, las células que sobreviven o que hacen apoptosis generan un estímulo antiinflamatorio y citoprotector, mientras que la necrosis es un importante estímulo proinflamatorio.
La segunda vía es favorecida con concentraciones altas de ATP asociadas a un aumento en la permeabilidad mitocondrial y a una concentración intermedia de ROS; los mediadores en esta vía son la caspasa 8-caspasa 3 y generan la respuesta de apoptosis45.
Por último, en situaciones con concentraciones elevadas de ROS, bajo ATP y una mayor permeabilidad mitocondrial, hay un aumento en la actividad del mediador RIP3 y se genera necrosis45 (fig. 2). El destino de cada célula es importante para el ambiente tisular, ya que la supervivencia celular y la apoptosis generan respuestas antiinflamatorias, mientras que la necrosis es un importante promotor de la inflamación46.
Además del efecto directo sobre la supervivencia celular, el TNF-α potencia la excitotoxicidad47,48, puede aumentar el flujo de calcio a través de la expresión de la subunidad GluR2 del receptor AMPA49, puede favorecer la liberación de más TNF, BDNF y glutamato por parte de las células microgliales50,51, en el astrocito —a ciertas concentraciones— bloquea la expresión del recapturador de glutamato GLT 1, siendo revertido el proceso al bloquear esta citocina o el BDNF52,53.
Interferon gamma (INF-γ)Esta citocina presenta también importantes funciones en el cerebro; por ejemplo, causa la polarización de las células del sistema inmune hacia M1, aumenta la expresión de los receptores para TNF y Fas, aumentando la sensibilidad a la apoptosis, y aumenta la actividad de enzimas encargadas de la producción de radicales libres como iNOS y la síntesis y liberación de quimiocinas54.
Entre las quimiocinas sobreexpresadas se encuentra IP 10 o CXCL10. Esta es un potente atrayente de linfocitos TH1 al tiempo que inhibe la migración y el cambio fenotípico a TH2; también aumenta la producción de INF-γ por parte de la microglía, generando una retroalimentación positiva55.
En modelos animales, la esplenectomía o el tratamiento con un anticuerpo policlonal contra INF redujo de manera significativa el tamaño del infarto y la recuperación funcional. Por otro lado, la IL-10 inhibe la producción de manera endógena de INF-γ, haciéndolo una terapia interesante54,55.
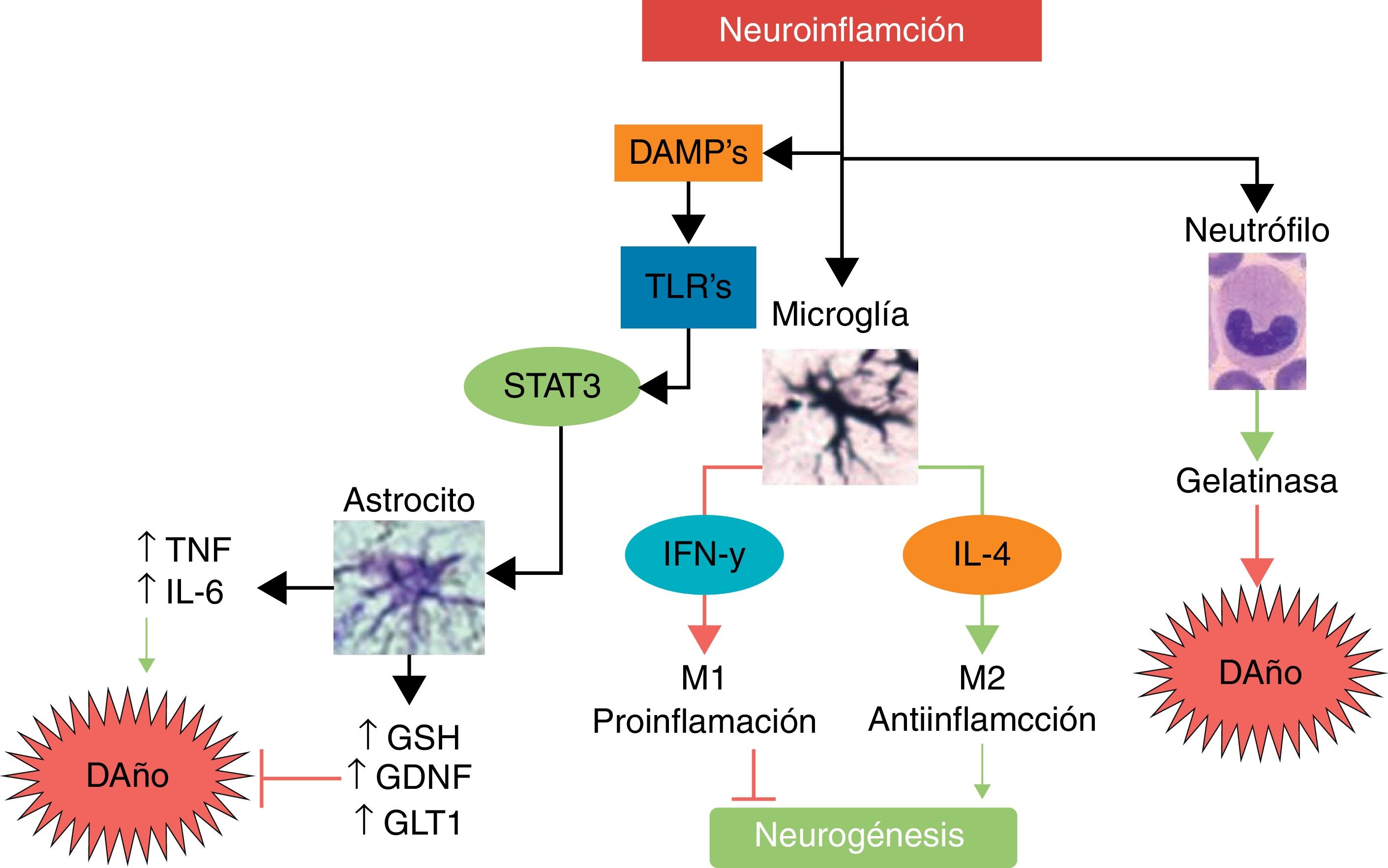
Interleucina 10 (IL-10)Esta es la principal citocina antiinflamatoria; como se ha mencionado antes, genera una polarización de las células inmunológicas al fenotipo M2, disminuye la migración de células proinflamatorias a través del endotelio, favorece la liberación de otras citocinas neuroprotectoras como IL-4, mientras que disminuye la secreción de TNF, IL-1, INF, y disminuye la producción de radicales libres y quimiocinas, entre otros efectos potencialmente protectores, tras un infarto cerebral56. En modelos animales de infarto cerebral, la IL-10 ha mostrado una importante disminución del volumen del infarto, así como menos déficits neurológicos57,58. En la figura 3 presentamos un resumen de algunas de las vías de daño y neuroprotección ante la neuroinflamación.

Principales mecanismos de daño y neuroprotección asociados a la neuroinflamación.La neuroinflamación no es un estímulo neurotóxico per se; al darse la activación inmunológica, las principales células que responden son las del sistema inmune innato tal como neutrófilos, microglía y astrocitos. Mientras que los neutrófilos son predominantemente neurotóxicos, la microglía y la astroglía pueden expresar fenotipos neuroprotectores. La microglía, dependiendo de la intensidad y del tipo de estímulo, puede cambiar a un fenotipo M1 con liberación de INF-γ, lo cual conlleva un mayor daño y una inhibición de la neurogénesis, mientras que el fenotipo M2 secreta IL-4, siendo antiinflamatorio y favoreciendo la neurogénesis y la neuroprotección. La astroglía, al ser activada por las moléculas de daño y dependiendo del receptor TLR que sea activado, puede secretar TNF e IL-6 favoreciendo el daño neuronal, o secretar factores neuroprotectores como el antioxidante GSH, el factor de crecimiento GDNF y el transportador GLT1.
Durante la isquemia se ha observado que la neurogénesis puede aumentar en el giro dentado del hipocampo, principalmente en los modelos de isquemia focal59-63. Sin embargo, aún no se ha establecido de manera determinante si esta activación tiene un rol en la recuperación de las funciones deterioradas por el evento isquémico. La proliferación y maduración de nuevas neuronas depende de diversos factores, entre los que destaca la inflamación64,65.
Los estudios muestran que la respuesta neurogénica puede modularse por la activación de la microglía en un evento isquémico, pero depende de si se encuentra en un estado proinflamatorio M1 o antiinflamatorio M2, inhibiéndola o favoreciéndola de manera respectiva66-69.
Estudios in vivo e in vitro mostraron que la microglía en sus estados 2 y 3 favorece la neurogénesis en GD del hipocampo a través de la secreción de TGF-β70 y IL-471. Además, la IL-15 tiene un rol importante en la zona subventricular de los ventrículos laterales al favorecer la proliferación de células progenitoras y su diferenciación neuronal72. Adicionalmente, la regulación de la IL-15 en un modelo de Alzheimer en ratones mostró que aumenta la neurogénesis in vivo y aumenta la proliferación de células progenitoras in vitro73. En cambio, factores como el TNF-α han mostrado que pueden inhibir la formación de nuevas neuronas en el hipocampo71,74,75. También se ha demostrado que la secreción de IL-1β disminuye la neurogénesis76,77, y la sobreexpresión de esta en ratones disminuye la diferenciación neuronal y sobrevivencia de nuevas neuronas78,79.
Recientemente se ha sugerido que el pretratamiento con antidepresivos, de la familia de los inhibidores selectivos de la recaptura de serotonina (ISRS), puede mejorar las alteraciones producidas por el evento isquémico en la clínica80, y en modelos de isquemia en animales81,82. Sin embargo el proceso no es claro. Algunas investigaciones sugieren que los ISRS como fluoxetina y citalopram inhiben la liberación de IL-1β, TNF-α y óxido nítrico por parte de la microglía activada, así como la liberación de glutamato y D-serina, favoreciendo la sobrevivencia de neuronas corticales en un modelo in vitro de hipoxia83. Este mismo efecto ha sido evaluado en un modelo in vitro de inflamación por administración de LPS, donde los ISRS también redujeron la liberación de TNF-α y óxido nítrico, sugiriendo un efecto antiinflamatorio84. Sin embargo, habrá que realizar más estudios para considerar seriamente los antidepresivos como estrategias terapéuticas ante un evento de isquemia cerebral.
ConclusionesNuestra compresión de las interacciones que presentan el cerebro y el sistema inmune crecen rápidamente, y con esto también las estrategias terapéuticas que podemos desarrollar para patologías tan relevantes como el ictus isquémico.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

