



La enfermedad de Alexander (AxD) es una leucodistrofia. Su base patológica, junto a la pérdida de mielina, es la aparición de los cuerpos de Rosenthal, que son inclusiones citoplasmáticas en células astrocitarias. Mutaciones en el gen que codifica la GFAP se han identificado como una base genética para AxD. Sin embargo, no se conoce el mecanismo por el cual estas variantes producen la enfermedad.
DesarrolloLa hipótesis más extendida es que AxD se desarrolla por un mecanismo por ganancia de función debido al incremento de GFAP. Sin embargo, este mecanismo no explica la pérdida mielínica, dado que los modelos experimentales que expresan GFAP normal o mutada no generan alteración mielínica. En la presente revisión se analizan otras posibilidades que permitan justificar dicha alteración, como son alteraciones epigenéticas, inflamatorias, la existencia de células NG2 (+)-GFAP (+) o cambios postraslacionales sobre la GFAP al margen de la mayor expresión.
ConclusionesLas diferentes hipótesis analizadas pueden explicar la alteración de la mielina que aparece en los pacientes y que pueden presentarse asociadas y abren la posibilidad de plantear terapéuticas basadas en estos mecanismos.
Alexander disease (AxD) is a type of leukodystrophy. Its pathological basis, along with myelin loss, is the appearance of Rosenthal bodies, which are cytoplasmic inclusions in astrocytes. Mutations in the gene coding for GFAP have been identified as a genetic basis for AxD. However, the mechanism by which these variants produce the disease is not understood.
DevelopmentThe most widespread hypothesis is that AxD develops when a gain of function mutation causes an increase in GFAP. However, this mechanism does not explain myelin loss, given that experimental models in which GFAP expression is normal or mutated do not exhibit myelin disorders. This review analyses other possibilities that may explain this alteration, such as epigenetic or inflammatory alterations, presence of NG2 (+) – GFAP (+) cells, or post-translational modifications in GFAP that are unrelated to increased expression.
ConclusionsThe different hypotheses analysed here may explain the myelin alteration affecting these patients, and multiple mechanisms may coexist. These theories raise the possibility of designing therapies based on these mechanisms.
La enfermedad de Alexander (AxD) es una leucodistrofia, descrita por el autor que lleva su nombre en 19491, que produce una alteración en la constitución de la mielina. Su base patológica, junto con la pérdida de mielina, es la aparición de los cuerpos de Rosenthal2, unas inclusiones citoplasmáticas en células de estirpe glial, que también han sido descritas en determinados gliomas. Estas inclusiones están constituidas por proteína glial fibrilar ácida (GFAP), α-β-cristalina y la proteína HSP273,4, aunque también se han hallado otras proteínas, como vimentina, p62 o plectina5. Desde el punto de vista clínico, la enfermedad tiene 3 formas, infantil, juvenil y adulta, siendo la primera de peor pronóstico6-8. Las mutaciones en el gen que codifica la GFAP se han identificado como una base genética para AxD. Estas mutaciones se basan en cambios en 32 nucleóticos específicos y aparecen tanto en casos familiares como esporádicos9,10. Sin embargo, no se conoce cómo estas mutaciones sobre el gen que codifica la GFAP pueden conducir a la agregación de la GFAP en los astrocitos del paciente, así como la forma en que la GFAP que expresan astrocitos contribuye a la clínica de los pacientes y especialmente por qué mecanismo se puede producir la pérdida mielínica. La pérdida mielínica es básica en la enfermedad, que presenta un cuadro radiológico característico con desmielinización periventricular11 que, en los casos de mayor supervivencia, llega a ser muy intensa, afectando a prácticamente toda la sustancia blanca12.
La GFAP, aislada y caracterizada por Eng en 196913, es un componente de los filamentos intermedios que se encuentra en los astrocitos junto con vimentina y nestina. Estos filamentos, aunque contribuyen en la estructura de los astrocitos y forman el citoesqueleto junto con los microtúbulos y los microfilamentos, tienen una función importante en la transmisión de señales. La GFAP también está presente en otras células del CNS, como son las células ependimarias, las células de Schwann que no producen mielina en el sistema nervioso periférico y las células de glía entérica, como parte del sistema nervioso entérico.
La GFAP está codificada por un solo gen que se halla en cromosoma humano 17q21, que tiene 9 exones. Existen al menos 10 isoformas que se generan como consecuencia de la selección alternativa de la unión de mRNA y la señal de poliadenilación14-17. La GFAP-α (isoforma 1) es la isoforma predominante en el cerebro y la médula espinal, pero también se presenta en SNP y tiene los clásicos 432 residuos con el pleno uso de los 9 exones. La GFAP-δ o GFAP-¿ (isoforma 2) se expresa preferentemente por astrocitos presentes en nichos neurogénicos, como en la zona subventricular y el hipocampo. La GFAP-δ incluye el uso de un intrón antes del exón-8 y tiene C-terminal alternativo y 431 residuos. La expresión de GFAP-δ se produce en astrocitos reactivos en patología, como en epilepsia, enfermedad de Alzheimer o gliomas. Las demás variantes son menos frecuentes, pero están recibiendo gran atención, porque algunas de ellas se han relacionado con enfermedades neurodegenerativas18,19.
La producción de mielina se realiza a partir de los oligodendrocitos (OL) y es un proceso dinámico que precisa de 3 cambios necesarios: que haya células precursoras de OL (OPC) presentes en el lugar desmielinizado, que se modifique su forma y que se produzcan cambios en su membrana y un microambiente adecuado. Las OPC son formas inmaduras de OL que tras el desarrollo embrionario aún permanecen en el cerebro adulto, constituyendo entre el 5 y el 8% de la población de células gliales en el SNC20 y contribuyen a la renovación de la vaina de mielina, diferenciándose a lo largo de la vida adulta. Estas células OPC pueden expresar algunas proteínas, como son Olig2 o NG2, que las diferencia. Para que exista una mielinización no deben existir unas condiciones que la impidan, como el hecho de que las propias proteínas mielínicas producidas y acumuladas como consecuencia de la desmielinización la impiden a través de la vías de señalización de la proteína cinasa C (PKC) α, del receptor Nogo 1 o la proteína que interactúa Lingo-1 (Nogo receptor interacting protein-1)21,22, así como las semaforinas que desempeñan también un papel importante en la regulación de la remielinización23. En consecuencia, para poder entender la pérdida mielínica que aparece en los pacientes con AxD, deberíamos considerar mecanismos que pudieran intervenir en al menos cuatro niveles24: 1) no se generan OPC o no sobreviven; 2) no existen estímulos que favorezcan la maduración de los OL; 3) existen factores inhibitorios locales que evitan la producción de mielina, o 4) existen alteraciones axonales que no son adecuadas para que el axón permita la mielinización. En esta revisión, se analizarán potenciales hipótesis que podrían justificar la alteración de la mielina en los pacientes con AxD.
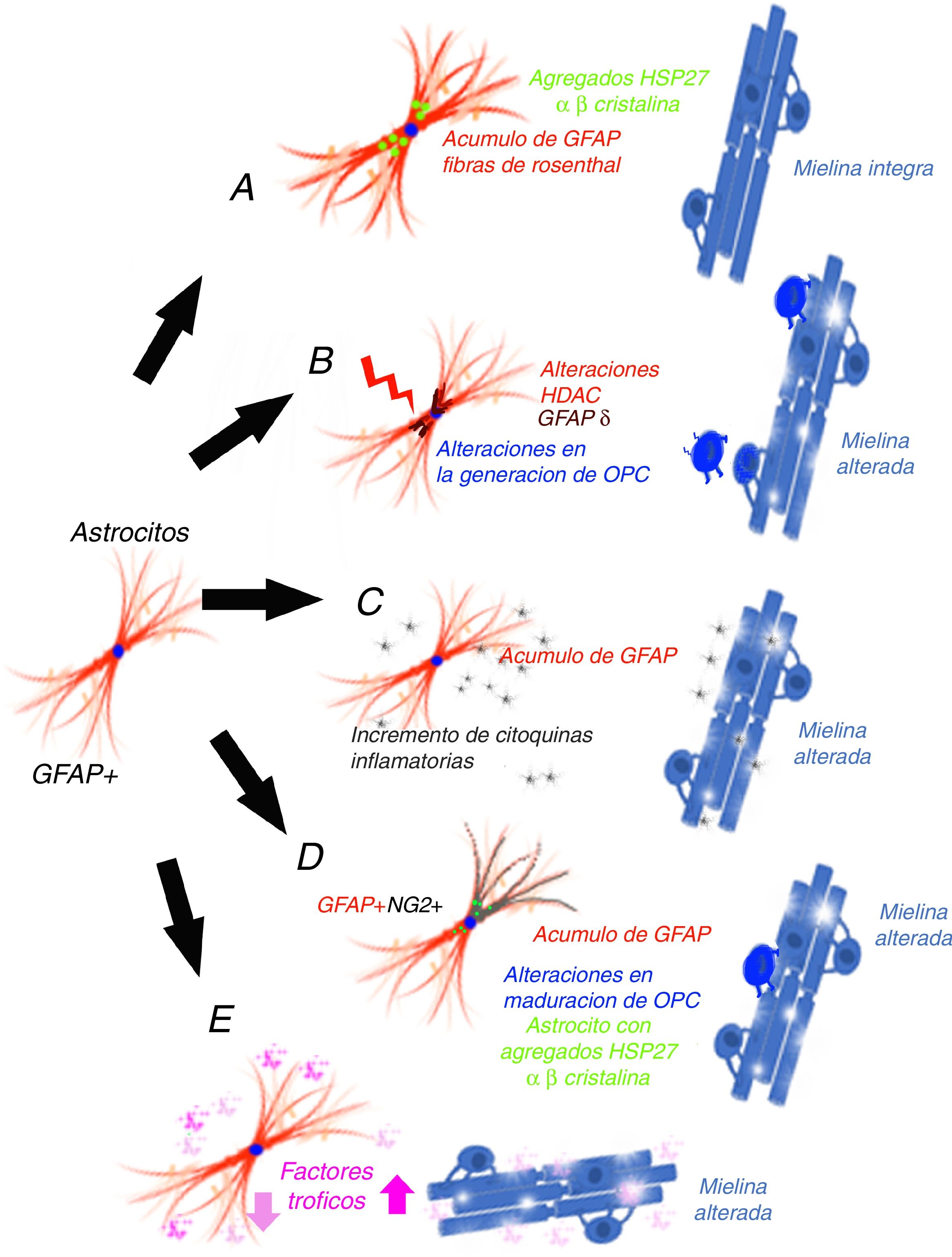
El mecanismo de ganancia de función de la proteína ácidica fibrilar glialMessing y su grupo han defendido que AxD se produce por el acúmulo de GFAP en astrocitos y que la elevación de la proteína es dañina, tanto la forma mutada como la no mutada25, por lo que han planteado un mecanismo de ganancia de función, similar al que se ha propuesto en otras enfermedades neurodegenerativas, como en las formas SOD1 dependientes de la esclerosis lateral amiotrófica26, aunque este mecanismo empieza a estar en discusión27. La GFAP se agregaría secuestrando las proteínas HSP 27, catepsina y α-β-cristalina, lo que tras una fosforilización y unión a ubiquitina generaría los cuerpos de Rosenthal y ello desencadenaría la lesión astrocitaria28, a la vez que la activación de vías de respuesta al estrés en estos astrocitos29,30, como la activación de la JMK y p3831. La disminución de la degradación de la GFAP puede estar relacionada con la disminución de la actividad proteosómica. Para estos autores32, dado que la GFAP tiene una vida media larga in vivo, interferiría en la degradación proteica de la célula de forma prolongada, lo que contribuiría aún más al acúmulo de GFAP33. Esta hipótesis ha generado un número muy importante de experimentos y modelos, especialmente en ratones que hiperexpresan tanto la GFAP mutada como normal. Tanaka et al.34 han publicado como la producción de un ratón transgénico que tiene cargada la mutación R239H. Messing y su grupo han descrito 2 modelos con ratones que reproduce las fibras de Rosenthal y el acumulo de proteína. A través de estos modelos se ha conseguido una importante información sobre qué mecanismos se desarrollan como consecuencia del acúmulo de GFAP35-40. Asimismo, se ha tratado de correlacionar el pronóstico de la enfermedad con la determinación de GFAP en el líquido cefalorraquídeo41. El principal problema de esta hipótesis es que no se genera desmielinización42 (fig. 1 A). Otra cuestión que limita esta hipótesis es que en los gliomas que presentan acúmulo de GFAP y que desarrollan fibras de Rosenthal tampoco hay perdida mielínica, de forma que existe una duda razonable sobre que el aumento de GFAP puede, por sí solo, explicar la enfermedad.
La alteración epigenética sobre la transcripción El mecanismo de ganancia de función de la GFAP. B) La alteración epigenética sobre la transcripción. C) El mecanismo inflamatorio. D) Las células GPAP (+)/NG2 (+). E) Las alteraciones postranscripcionales de la GFAP.")
Los niveles de expresión de GFAP están controlados por la actividad del promotor del gen de GFAP, un proceso que depende en gran medida de modificaciones epigenéticas. Durante la iniciación de la astrogénesis en células madre neurales, la desmetilación del promotor de GFAP activa la transcripción de GFAP43-46, de forma que la acetilación de histonas controla la expresión de GFAP en la diferenciación de las NSC, dependiendo del estado de diferenciación de la célula47,48. El estado de acetilación de las proteínas histonas está regulado por las enzimas histona acetilasa e histona desacetilasa (HDAC). Kanski et al.49 han demostrado que la acetilación de histonas en astrocitos es un importante regulador de la transcripción, así como del splicing alternativo de GFAP. La inhibición de HDAC reduce significativamente la expresión de GFAP en astrocitos humanos primarios, así como en células de astrocitoma. Este mecanismo es relevante, ya que la inhibición de HDAC modifica la proporción entre la transcripción alternativa GFAP-δ y la isofórmica constitutiva GFAP-α a favor de la expresión de GFAP-δ, lo que se modifica durante la diferenciación hacia células de estirpe astrocitaria o oligodendrocítica. En este sentido, la inhibición de la actividad HDAC modifica la estructura de los filamentos de GFAP en la célula, tanto en su extensión, localización y grado de agregación. Diferentes estudios han mostrado que las modificaciones en la agregación de la GFAP pueden asociarse a alteraciones en la diferenciación en leucodistrófias50-52, asociándose a diferencias en la expresión de GFAP-δ. Un paciente con AxD, con una mutación en el gen GFAP y una mutación en el gen HDAC6, se asoció con un fenotipo más grave de la enfermedad53 y con una actividad reducida de HDAC6. La hipótesis de estos autores permitiría, en consecuencia, explicar como en la AxD existiría en acúmulo de GFAP y alteraciones en la diferenciación oligodendrocitaria que explicarían la pérdida de mielina (fig. 1 B).
El mecanismo inflamatorioOlabarria et al. han indicado que AxD podría estar mediada por un mecanismo inflamatorio54. En la esclerosis múltiple y en la neuromielitis óptica existe una desmielinización y su origen es inmunitario, y atribuido en parte a mecanismos inflamatorios y al incremento de citocinas proinflamatorias. Kondo et al.55 han estudiado células madre pluripotentes inducidas (iPSC), obtenidas de 3 pacientes AxD, con diferentes mutaciones del gen de la GFAP. Los astrocitos derivados de AxD iPSC mostraron agregados citoplasmáticos positivos a GFAP, como las fibras de Rosenthal, pero también mostraron una liberación alterada de citocinas. Algunos artículos han descrito la infiltración linfocítica o la activación microglial en el cerebro de pacientes con AxD56,57, aunque no de forma excesivamente importante. Estos autores han estudiado la expresión de citocinas en modelo de ratones por sobrexpresión de GFAP y en ratones heterocigotos para la mutación R236H de GFAP, detectando la respuesta inflamatoria. Las iPSC procedentes de los pacientes con AxD mostraron un incremento de citocinas proinflamatorias, como GM-CSF, IL-5, IL-6 y factor de necrosis tumoral-α. Asimismo, se ha señalado que la molécula de GFAP puede ser deaminada58 y, en ese caso, se ha observado que podría evocar una respuesta autoinmune59. Por todo ello, estos autores han indicado que un proceso neuroinflamatorio puede estar involucrado en la patogénesis de AxD y que la pérdida mielínica podría tener un mecanismo inmunitario, como ocurre en la esclerosis múltiple (fig. 1 C).
Las células GPAP (+)/NG2 (+)Las células gliales NG2 (+)60,61, también llamadas sinantocitos62 o polidendrocitos63, representan el 8-9% de las células en la sustancia blanca y el 2-3% de las células de la sustancia gris64, y fueron inicialmente identificadas como células progenitoras Olig 265, ya que se diferenciaban en OL66-70. En realidad, son OPC capaces de recibir entrada sináptica que puede regular su destino celular. Estas células también pueden extender los procesos al nodo de Ranvier y pueden transformarse en astrocitos reactivos en condiciones patológicas y en cultivos celulares71,72. Representan, por tanto, un estadio de transición entre los OL y los astrocitos73. Existen células NG-2 (+) con GFAP (+) en determinadas situaciones patológicas, como en el área que rodea la desmielinización en esclerosis múltiple74, y también se han descrito la presencia de GFAP (+) en células de estirpe oligodendrocitaria en leucodistrofias, alteraciones mielínicas de origen viral o inmunitario75-77. En el ratón que hiperexpresa GFAP mutada, se han hallado alteraciones en la neurogénesis hipocámpica78. El grupo del que forman parte los autores de este artículo han coincidido en este hallazgo en un modelo celular tras la transfección de mutaciones de AxD79. La expresión de GFAP en las células de Schwann genera que estas células se comporten funcionalmente como astrocitos y no generen mielina80. La expresión de GFAP se ha hallado en células del linaje oligodendroglial en fases iniciales y la expresión de NG2 en células que se diferenciarán a astrocitos81-84, y la coexistencia de GFAP (+)/NG-2 (+) en células hace muy difícil señalar si es un OL o un astrocito. El trasplante de células OPC en el SNC sin glía genera simultáneamente OL y astrocitos85. La persistencia de GFAP (+) en células que deberían diferenciarse hacia la estirpe oligodendrocitaria justificaría el acúmulo de la proteína y la ausencia de generación de mielina (fig. 1 D). Es muy probable que la persistencia de GFAP (+) en células NG2 (+) y, en consecuencia, en la dirección a una estirpe pueda depender de la isoforma de GFAP o de factores relacionados con el microambiente en que se desarrolla el proceso de maduración.
Las alteraciones postranscripcionales de la GFAPLos astrocitos son células altamente secretoras de factores tróficos86, algunos de ellos influyen en la mielinización. En algunos modelos experimentales se ha demostrado que BDNF influye en la proliferación y remielinización87,88, y es posible que la interacción astrocito-OL sea esencial en la generación de mielina89,90. Los experimentos de cultivo celular proporcionan evidencia directa de que el BDNF astrocítico promueve la maduración de OPC y los datos in vivo muestran que los ratones transgénicos con baja regulación de BDNF en astrocitos presentan menos oligodendrogénesis. Otros factores de crecimiento, como CNTF,91 FGF, TGF β o GDNF, así como receptores nucleares, pueden modificar la activación de la transcripción del gen de la GFAP92. Yang y Wang han revisado exhaustivamente y han llamado la atención de la complejidad de los mecanismos que se desarrollan tras la transcripción de la GFAP93. La GFAP está sometida a una serie de modificaciones postraduccionales, altamente regulada por las proteínas cinasas, de forma que modificaciones específicas sobre la propia molécula, debidas a cambios genéticos o al microambiente, pueden influir en función de los astrocitos y podrían intervenir en la mielinización (fig. 1 E).
ConclusiónEl mecanismo de ganancia de función permitía albergar esperanzas del hallazgo de un fármaco que, descendiendo la expresión de GFAP, produjera la mejora de los pacientes. En este sentido, la búsqueda de una terapéutica potencialmente efectiva, a través de modelos celulares o en los ratones que hiperexpresan GFAP, ha sido intensa94,95. La cuestión básica es que si los cambios producidos por el aumento de GFAP, aunque sean deletéreos, son secundarios, estas terapéuticas serían sintomáticas, y en una enfermedad que probablemente se inicia durante el desarrollo o en el periodo neonatal serían insuficientes.
Las diferentes hipótesis planteadas pueden explicar la alteración de la mielina que aparece en los pacientes y que debe ser relevante y algunas de ellas pueden presentarse relacionadas. La posibilidad de utilizar terapéuticas que modifiquen los mecanismos epigenéticos96 parece muy razonable y de enorme vigencia, y ya se están realizando ensayos clínicos en el cáncer97. Asimismo, la actuación sobre la diferenciación de las células inmaduras hacia las estirpes astrocitaria y oligodendrocitaria podría plantearse como una línea potencialmente terapéutica.
Conflicto de interesesLos autores declaran que este artículo se ha realizado en ausencia de relaciones comerciales o financieras que pudieran interpretarse como un posible conflicto de intereses.
A la Fundación Ayuda-Juanma (www.ayudajuanma.es), por el patrocinio económico de la línea de investigación sobre AxD en el Hospital Clínico San Carlos.

