




La presencia simultánea de rigidez meníngea y lesiones cutáneas, alerta al clínico respecto a una probable etiología infecciosa. Sin embargo, existen causas alternativas, como las secundarias a fármacos, neoplasias o enfermedades inflamatorias, figurando entre estas, el síndrome de Sweet (SS).
El SS o dermatosis neutrofílica febril aguda, es una entidad de etiología desconocida, que puede aparecer aislada, asociada a neoplasias, o a enfermedades inflamatorias y autoinmunes. El SS presenta lesiones cutáneas, clínica sistémica y, en ocasiones, síntomas neurológicos.
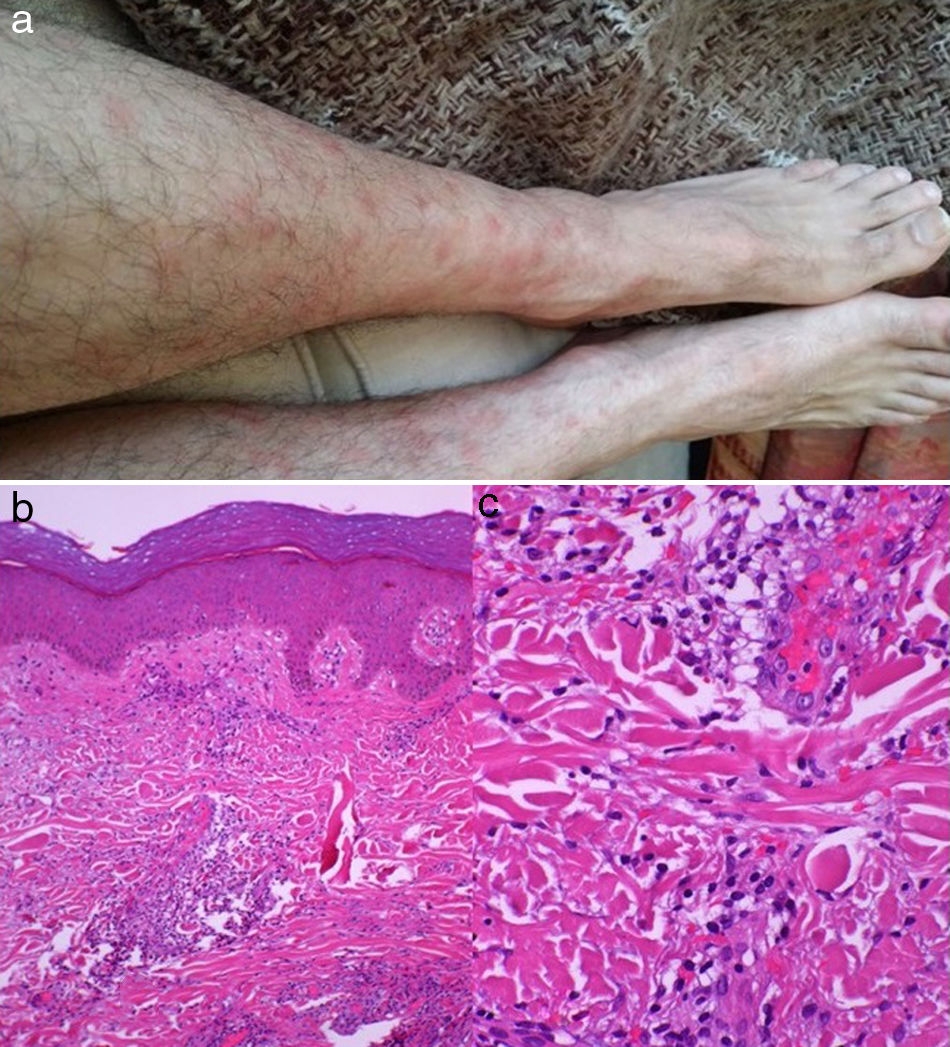
Se describe el caso de un varón de 47 años, sin antecedentes de interés, con historia de 2 semanas de malestar, odinofagia, cefalea holocraneal, osteomialgias y fiebre, que recibió amoxicilina e ibuprofeno, sin mejoría. Al cabo de una semana, presentó rigidez de nuca y una erupción cutánea dolorosa, en forma de placas y nódulos eritematosos confluyentes, en piernas, brazos y tórax.
Entre las pruebas complementarias, en el líquido cefalorraquídeo (LCR) se evidenció una pleocitosis de 73 células/mm3 (100% linfocitos); hiperproteinorraquia (0,53g/l) y glucorraquia normal. Los cultivos bacterianos y la reacción de la polimerasa en cadena para herpesvirus, enterovirus, Streptococcus, Listeria monocytogenes y Neisseria meningitidis, fueron negativos. La analítica sanguínea mostró una velocidad de sedimentación globular de 94mm/h; proteína C reactiva de 4,82mg/dl (normal: ≤0,5mg/dl), leucocitosis (12.700/mm3, 78% neutrófilos); IgM anticardiolipina 21,91U/ml (normal: ≤12,5U/ml); IgM anti-β2glicoproteína 1 (B2GP1) 104,51U/ml (normal: ≤20U/ml), y marcadores tumorales normales. Una biopsia cutánea demostró un infiltrado constituido por neutrófilos en la dermis, sin afectación de las paredes vasculares, ni microtrombosis (fig. 1). Una TAC toraco-abdominal y una gastroscopia resultaron normales, así como una RM craneal. No se halló asociación con HLA CW1, ni con B54.
. Biopsia cutánea: Tinción con hematoxilina y eosina, se evidencia un infiltrado neutrofílico intradérmico sin afectación vascular ni microtrombos (b y c).")
Se administró ceftriaxona y aciclovir, que se suspendieron ante la ausencia de infección bacteriana o viral. Se inició, entonces, tratamiento con metilprednisolona oral, 75mg/día, mejorando la erupción cutánea y la clínica neurológica. Seis meses después, los anticuerpos IgM e IgG anti-B2GP1 resultaron negativos.
Ante la ausencia de afectación vascular en la biopsia cutánea y la respuesta espectacular al tratamiento con corticoides, una vez suspendidos los antimicrobianos y descartadas etiologías alternativas, el presente caso fue catalogado de SS con afectación neurológica (NS).
El SS es una enfermedad inflamatoria infrecuente, que puede presentar una amplia variedad de manifestaciones. Se caracteriza por inicio abrupto de edema y lesiones eritematosas dolorosas en la piel, en forma de placas, nódulos o pápulas, así como fiebre y leucocitosis con neutrofilia1. Puede afectar a distintos órganos aparte de la piel, como los sistemas respiratorio, musculoesquelético y nervioso central (SNC).
Histopatológicamente, se halla un infiltrado inflamatorio compuesto por neutrófilos en la dermis, sin invasión vascular ni evidencia de infección. Ocasionalmente, se dan variantes en que la celularidad es de predominio mononuclear con apariencia de histiocitos, que en realidad son granulocitos inmaduros (SS histiocitoide)2.
El SS se ha relacionado con una variedad de enfermedades, en función de lo que se clasifica como: 1) SS clásico; 2) asociado a neoplasia; y 3) inducido por fármacos.
El SS clásico representa la mayoría de los casos, y se denomina de esta forma, cuando se descarta asociación con tumores malignos o fármacos. El SS clásico puede vincularse a infecciones, enfermedad inflamatoria intestinal, embarazo y enfermedades autoinmunes3.
Cohen et al. describieron 79 pacientes con SS asociado a neoplasia, de los que el 87% se asoció a tumores hemáticos (linfoma, leucemia crónica, mieloma y síndromes mielodisplásicos), y el resto, a tumores sólidos4. En otro estudio, estos mismos autores describieron 118 casos de SS, en los que el 21% se relacionaron con tumores sólidos o hematológicos5.
El SS se ha relacionado con múltiples fármacos, entre los que el más frecuente fue el factor estimulante de colonias de granulocitos (tabla 1)3.
Fármacos asociados al síndrome de Sweet
| Antibióticos | Clindamicina, ciclinas (doxiciclina, minociclina y tetraciclina), nitrofurantoína, quinolonas (norfloxacina y ofloxacina), piperacilina/tazobactam, estreptograminas (dalfopristina/quinupristina), trimetoprim-sulfametoxazol |
| Antivirales | Abacavir, aciclovir, interferón-α |
| Bioterapéuticos | Inhibidores de proteosomas (bortezomib), inhibidores de la tirosina-quinasa (imatinib, nilotinib), factores estimulantes de colonias de granulocitos, ácido transretinoico, factor estimulante de colonias de macrófagos |
| AINE | Celecoxib, rofecoxib, diclofenaco |
| Psicotropos | Amoxapina, clozapina, diazepam, lormetazepam |
| Vacunas | Bacilo de Calmette-Guérin, Streptococcus pneumonia (Pneumovax®), influenza, viruela |
| Miscelánea | Azatioprina, agentes de contraste radiológico, propiltiouracilo, carbamazepina, cloroquina, furosemida, hidralazina, isotretinoína, lenalidomida, anticonceptivos orales |
La patogenia del SS es desconocida. Sin embargo, se han propuesto diversas teorías sobre ella, entre las que destacan una predisposición genética en los portadores de HLA, CW1 y B54, disregulación de citoquinas y diversas reacciones de hipersensibilidad.
La afectación del SNC en el SS, en forma de meningitis o encefalitis, además de otras complicaciones neurológicas, ha sido ampliamente cubierta en la literatura médica. Hisanaga et al.6, describieron los hallazgos en 42 casos de NS, y propusieron criterios diagnósticos para la afectación neurológica en el SS.
Un caso de probable NS debería cumplir los siguientes criterios: 1) síntomas neurológicos que respondan rápidamente a corticoides; 2) placas o nódulos dolorosos localizados en cara, cuello, tronco o extremidades superiores, con un infiltrado neutrofílico en la dermis y ausencia de vasculitis; 3) ausencia de uveítis o vasculitis cutánea (a diferencia del síndrome de Behçet), y 4) exclusión de otras causas de afectación del SNC. Como puede verse, nuestro caso cumple los criterios diagnósticos de esta categoría.
El pronóstico de NS es generalmente bueno, con respuesta favorable a los corticoides sistémicos, aunque puede haber recurrencias7. El diagnóstico diferencial debe incluir cualquier causa de meningoencefalitis linfocitaria, especialmente el síndrome de Behçet8.
Los anticuerpos antifosfolípido se han vinculado a numerosas enfermedades autoinmunes y están presentes hasta en el 5% de la población, si bien solo una pequeña parte desarrolla complicaciones vasculares9. La asociación de anticuerpos anti-B2GP1 y el SS no ha sido descrita previamente en la literatura médica, siendo el objetivo de este trabajo, comunicar dicha asociación.
Por otra parte, la desaparición de los anticuerpos IgM anti-B2GP1, junto a la ausencia de conversión a IgG de estos anticuerpos, la resolución de las manifestaciones cutáneas y neurológicas tras el tratamiento corticoideo y la ausencia de fenómenos vasculares o neoplásicos tras 18 meses de seguimiento, apoyarían la posibilidad de que este SS clásico con manifestaciones neurológicas, fuera de etiología autoinmune.
Presentado como poster en la LXVI reunión anual de la SEN en Valencia, noviembre de 2014.

