



La acidemia metilmalónica con homocistinuria, es un error congénito poco frecuente del metabolismo de la vitamina B12 (cobalamina). Está causada por defectos en la síntesis de los coenzimas adenosilcobalamina (AdoCbl) y metilcobalamina (MeCbl) que provocan disminución en la actividad de los enzimas respectivos metilmalonil-CoA mutasa (MUT;609058)y metiltetrahidrofolato:homocisteína metiltransferasa, también conocida como metionina sintasa (MTR;156570) Existen 4 grupos de complementación por defectos de la cobalamina, asociados a la acidemia metilmalónica con homocistinuria (cblC, cblD, cblF y cblJ) causadas por mutaciones en los genes implicados. La acidemia metilmalónica con homocistinuria tipo cblD está causada por mutaciones en el gen MMADHC (2q23.2) que se transmite siguiendo un patrón autosómico recesivo. A continuación describimos dos casos.
Paciente 1. Mujer de 18 años de etnia gitana. Sin antecedentes de interés, realiza una dieta alimenticia sin control e ingresa por fiebre de 38,5°C, junto con cuadro confusional brusco, desorientación temporo-espacial, lenguaje incoherente, no disártrico y lentitud psicomotora. Presenta buen nivel de consciencia, alerta, desorientada, inquieta e irritable, sin agresividad. Sin cefalea o signos meníngeos. Hemograma, bioquímica, proteinograma, inmunoglobulinas, autoinmunidad, serología, hormonas tiroideas, ferritina, vitamina B12 y ácido fólico normales. La tomografía computarizada craneal y la punción lumbar resultaron normales (estudio microbiológico negativo).
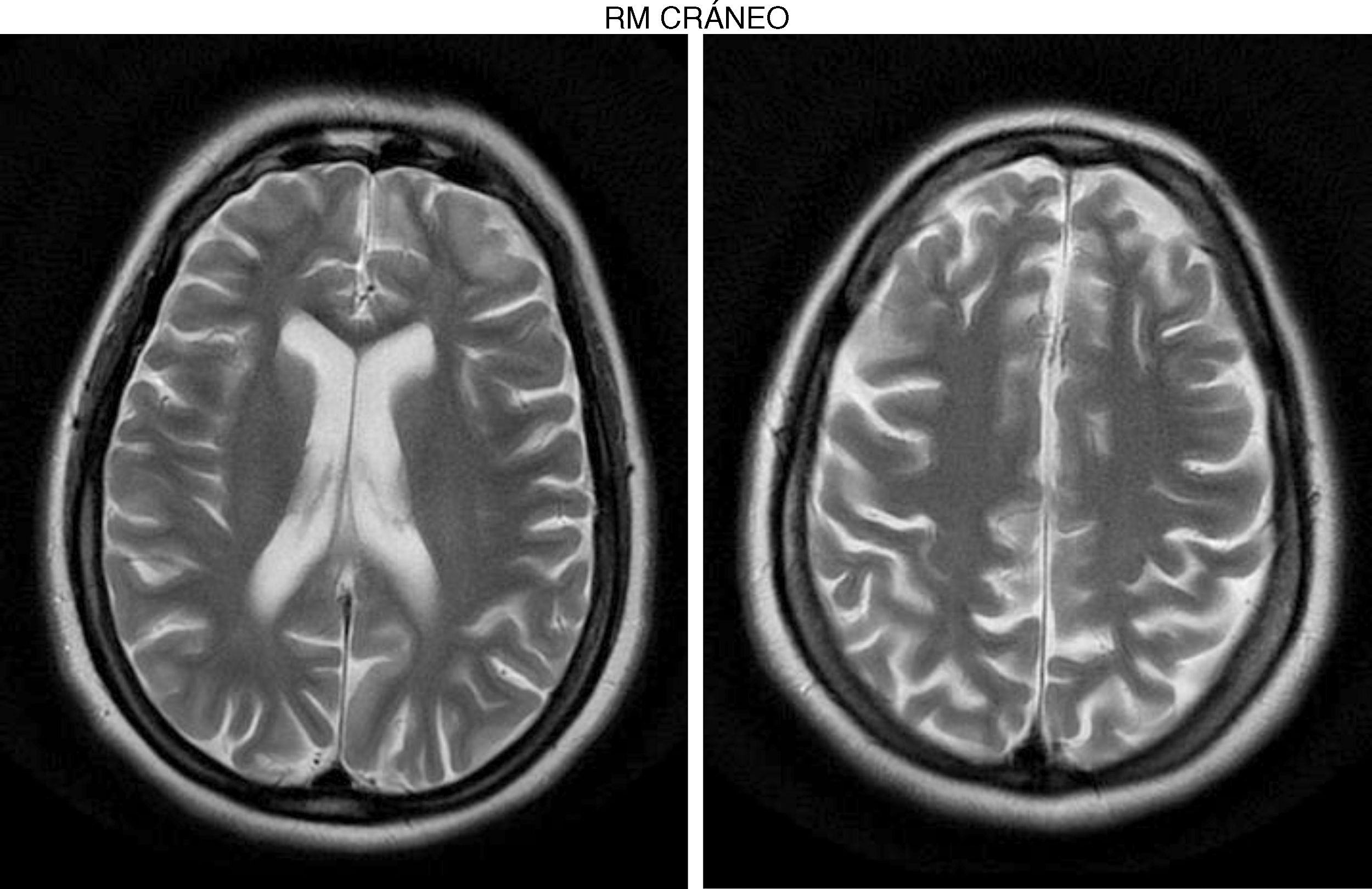
Recibió tratamiento empírico por Medicina Interna con aciclovir, presentando una evolución favorable y desapareciendo completamente la sintomatología. No acude a las revisiones posteriores programadas al alta y 10 meses más tarde consulta por trastorno gradual y progresivo de la marcha (debilidad en miembros inferiores), caídas frecuentes, con pérdida de fuerza en miembros superiores. En la exploración se encuentra alerta, con leve disartria, bradipsiquia muy marcada y dificultad para mantener la atención. Los reflejos de liberación frontal eran positivos. Las pupilas eran isocóricas normorreactivas. La exploración muscular muestra una fuerza muscular en el psoas 3/5, cuádriceps 3/5, glúteos medianos 4/5, glúteo mayor 3/5, tibial anterior 2/5, extensores de los dedos 1/5, isquiotibiales 3/5, dorso flexor del tobillo 1/5 y tibial anterior 2/5. Existe hiporrreflexia rotuliana 2/4, además de clonus aquíleo bilateral. Miembros superiores: reflejos 3/4 y fuerza 4/5. Sensibilidad profunda distal afectada e hipoestesia. Ataxia en la marcha y espasticidad de grado i. Babinski positivo bilateral. La RM craneal realizada (fig. 1) muestra retracción cortical, ligera ampliación de espacios subaracnoideos de la convexidad y discreta dilatación del sistema ventricular supratentorial con adelgazamiento del cuerpo calloso. El ECG asimismo mostró una lentificación del trazado de fondo sin anomalías epileptiformes. El electroneurograma muestra una discreta polineuropatía axonal sensitivomotora de predominio en miembros inferiores. Se realiza un diagnóstico diferencial incluyendo estudio de paraparesias espásticas genéticas familiares que resultó negativo. Asimismo el cribado de enfermedades autoinmunes y paraneoplásicas también resultó negativo. Se decide entonces ampliar el estudio hacia errores innatos del metabolismo de comienzo tardío, solicitando niveles de homocisteína en sangre, aminoácidos y ácidos orgánicos en sangre y orina de 24 h, detectándose entonces un aumento de ácido metilmalónico (258 nmol/mol de creatinina, normal<0,56) en orina, de ácido metilcítrico y ligera cetosis en orina (ácido 3-hidroxibutírico), así como homocisteína en sangre elevada (339,2μmol/l, normal < 10). Durante el estudio, la paciente presenta una trombosis venosa poplítea, con tromboembolismo pulmonar bilateral masivo que causa su muerte.

Paciente 2. Mujer de 15 años de etnia gitana. Hermana de la paciente 1, 3 años menor.
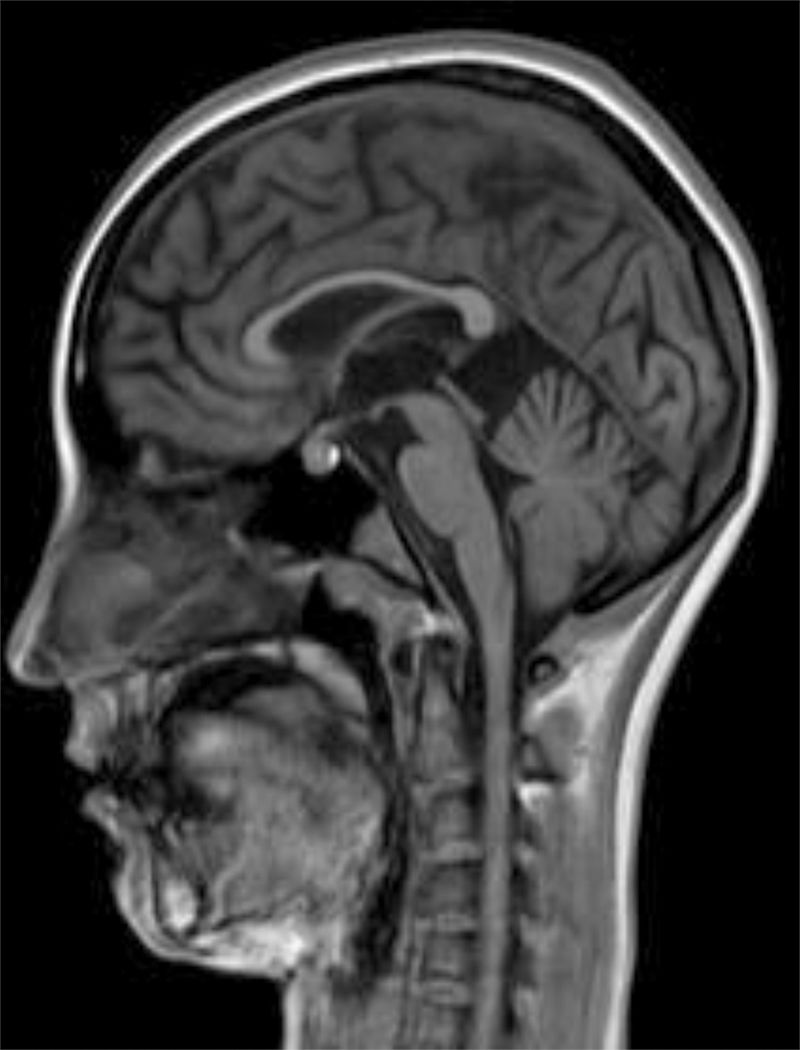
Desarrollo previo normal. Pocos meses antes de fallecer su hermana, inicia un cuadro insidioso de dificultad en la marcha, pérdida de apetito y risa inapropiada. En la exploración, destacan bradipsiquia y dificultad para mantener la atención, marcha atáxica con espasticidad grado i global, aproximación de caderas y leve recurvatum de rodillas, balance muscular del psoas 4/5, cuádriceps 3/5, flexión dorsal 1/5, clonus inagotable en sóleos en sedestación, agotable en supino, leve ataxia en MSI. Aumento simétrico de reflejos osteotendinosos en miembros inferiores. Babinski bilateral. El balance muscular en miembros superiores de 4/5 proximal y 5/5 distal. Fondo de ojo normal y RM muy similar a su hermana (fig. 2). El ECG muestra lentificación del trazado de fondo. Presenta 2 crisis epilépticas generalizadas tonicoclónicas sin proceso desencadenante, que requieren tratamiento antiepiléptico.

El estudio de aminoácidos y ácidos orgánicos muestra un aumento de homocisteína en sangre (169,9μmol/l, normal < 10) y ácido metilmalónico en orina (15,40 nmol/mol de creatinina, normal < 0,56). El estudio de genes implicados en la AM con homocistinuria muestra mutación homocigótica c.748C>T en el gen MMADHC (comprobado en el ADN de ambas hermanas), que genera un codón de stop dando lugar a una proteína truncada causante de la patología1–3. La enferma recibe tratamiento actualmente, consiguiéndose una sustancial mejoría y estabilización neurológica.
ConclusiónEl diagnóstico definitivo de estos pacientes permite la aplicación precoz del tratamiento específico, así como el consejo genético y el diagnóstico prenatal si se requiere.
Consideramos que ante sintomatología como paraparesias, polineuropatias axonales y desmielinizantes y deterioro cognitivo de causa no explicada, así como trombosis venosas profundas, deben estudiarse la homocisteína y el metabolismo de la vitamina B124,5.
Consentimiento informadoEl artículo presente es enviado para su publicación con el consentimiento por escrito de los padres de ambas pacientes.
Contribución de los autoresDra. E. Cancho García. Se encarga del diagnóstico clínico y tratamiento. Dra. E. Geán y Dr. T. Oliver Tormo, se enacrgan del estudio genético-metabólico. Dr. Torrents, se encarga del estudio metabólico. Dra. E. Esteban Durán, se encarga de proporcionar las imágenes radiológicas.

