



Presentamos la exploración oftalmológica de una paciente diagnosticada de un síndrome de epilepsia mioclónica y fibras rojas rasgadas (MERRF) asociado a la enfermedad de Madelung (lipomatosis simétrica múltiple) y portadora de la mutación mitocondrial primaria T14484C relacionada con la neuropatía óptica de Leber (LHON)1.
La enfermedad de Madelung es una entidad rara (1:25.000) caracterizada por múltiples lipomas no encapsulados distribuidos simétricamente en torno al cuello y la cintura escapular2. Pueden aparecer otras neuropatías, miopatías o afectación del sistema nervioso autónomo2. Su etiología es incierta3, pero la mayoría de los casos se asocian con alcoholismo4, trasplantes hepáticos5, enfermedades endocrinológicas y metabólicas (diabetes mellitus, hiperuricemia e hipercolesterolemia)6. En el 10% de los casos aparece la mutación del ADN mitocondrial m8344A>G relacionada con el síndrome MERRF7,8.
Caso clínicoPresentamos el caso de una mujer de 63 años sin hábitos tóxicos, con lipomas cervicales secundarios al síndrome de Madelung, síndrome de Wolf Parkinson White (WPW), polineuropatías y mioclonías, cuyo cuadro sistémico ya fue descrito por López-Blanco et al.1. Su madre presentaba temblor, y tanto ella como su hermana, lipomas.
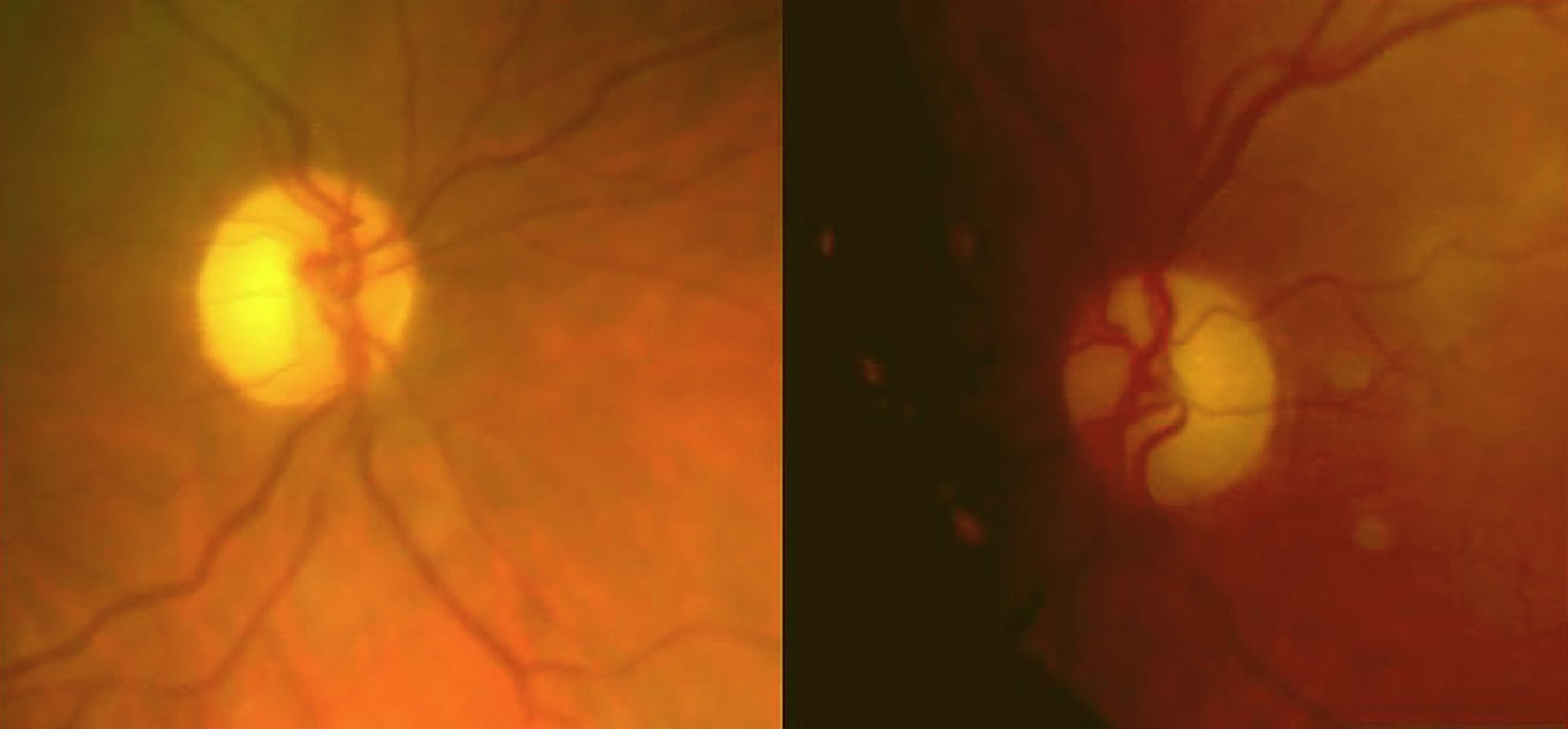
Refería una disminución progresiva e indolora de la agudeza visual (AV) del ojo derecho (OD) de un año de evolución (AV OD: 0,7; AV OI: 1). Se observó una palidez bitemporal de ambos nervios ópticos (fig. 1).

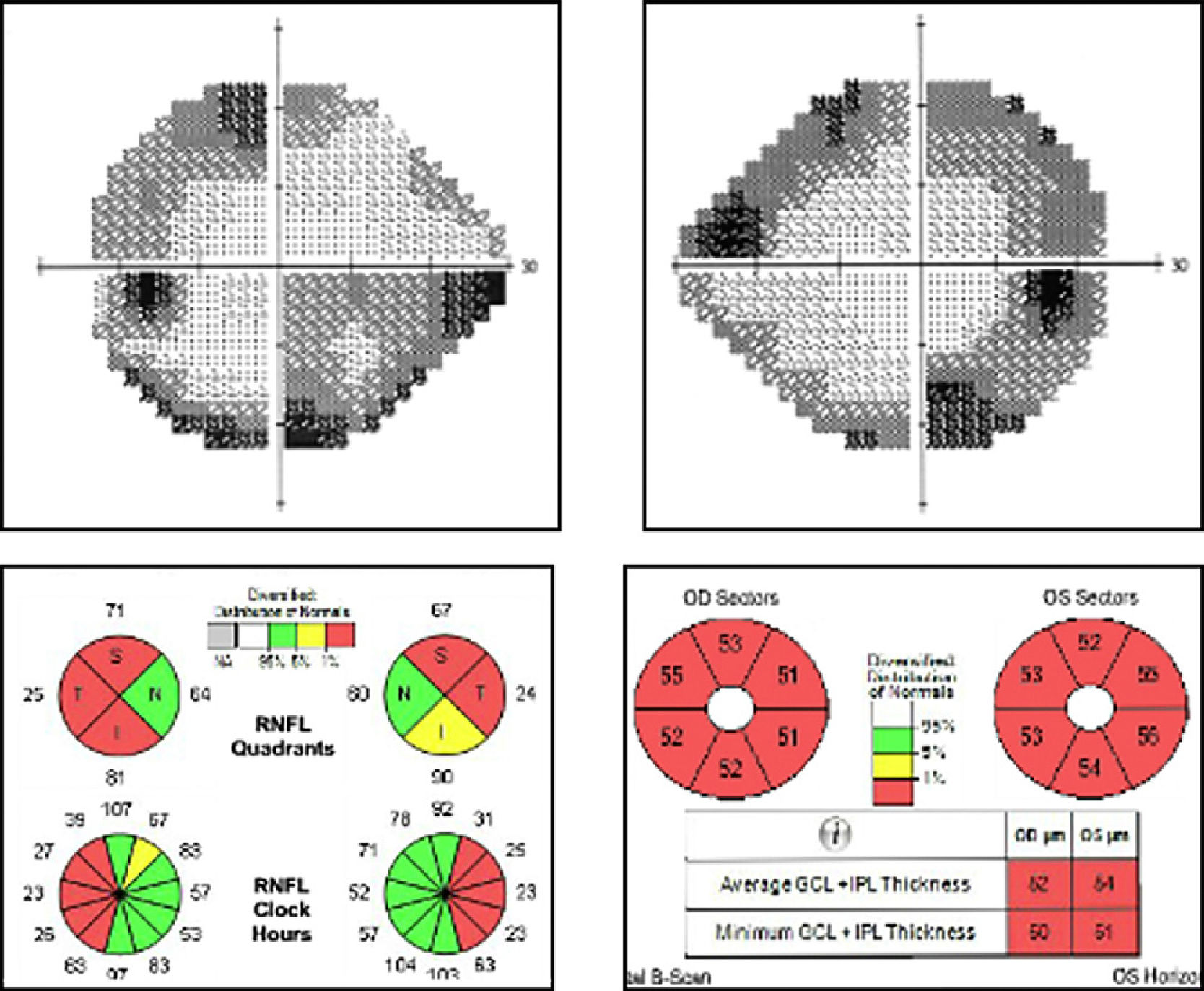
El campo visual mostró defectos concéntricos en ambos ojos (AO) y disminución de la sensibilidad general, sin afectación de los puntos centrales (VFI y DM de 80% y –11.20dB en el OD y de 75% y –11.21dB en el OI). Distinguía 2 de 12 de las láminas de Ishihara. La tomografía de coherencia óptica reveló una disminución de la capa de fibras nerviosas de predominio temporal (medias en AO de 60μ), y pérdida generalizada de las células ganglionares (medias de 52μ en OD y de 54μ en OI) (fig. 2). El electrorretinograma (ERG) multifocal era normal y la onda N95 estaba alterada en el ERG patrón.

La resonancia magnética nuclear cerebral mostró atrofia supra e infratemporal sin engrosamiento de los nervios ópticos en T2. El electromiograma y el electrocardiograma apoyaron los diagnósticos de polineuropatía y síndrome de WPW.
El análisis genético mostró 2 mutaciones del ADN mitocondrial: m8344A>G y T14484C. La biopsia muscular confirmó la presencia de fibras rojo rasgadas.
Por todo ello, la paciente fue diagnosticada de síndrome MERRF junto con enfermedad de Madelung y portadora de una de las mutaciones primarias del LHON.
Se aconsejó evitar fármacos tóxicos para la cadena respiratoria y comenzar tratamiento con coenzima Q10.
Durante el seguimiento (2 años) la agudeza visual se ha mantenido estable.
DiscusiónEste caso muestra un curioso solapamiento de 2 mutaciones mitocondriales en el mismo individuo, que repercuten en varios órganos con elevado requerimiento energético9 (corazón, músculos esqueléticos, cerebro y sistema visual, en concreto, haz papilomacular)10.
Las mutaciones que causan MERRF dificultan la cadena respiratoria mitocondrial y la síntesis proteica. Son características las mioclonías, la mielopatía y la espasticidad, junto con las neuropatías periféricas. Los pacientes suelen ser de estatura corta y raramente tienen lipomas. También pueden presentar ataxia, sordera, demencia y, en algunos casos, epilepsia. Algunos casos asocian atrofia óptica y retinopatía11,12.
Teniendo en cuenta que nuestra paciente presenta 2 potenciales mutaciones causantes del déficit visual (m8344A>G y T14484C), que respectivamente producen cuadros oftalmológicos en un 20% de los casos, consideramos la primera como la principal responsable del mismo, y no la relacionada con el LHON por las siguientes razones: la epidemiología (era mujer) y la patocronia (pérdida visual unilateral y progresiva) no son típicas del LHON; la visión central estaba relativamente conservada y el defecto campimétrico sin afectación central tampoco son sugestivos del LHON13. El ERG patrón nos muestra una alteración de la onda N95 con normalidad de la P50 lo que sugiere una alteración de las células ganglionares14. En el LHON es característica la microangiopatía discal (pseudoedema, hiperemia, telangiectasias peripapilares y edema de la capa de fibras nerviosas retinianas) y la atrofia óptica tardía; además en la resonancia magnética nuclear el nervio óptico puede estar engrosado en T2. El WPW de nuestra paciente podría relacionarse con la mutación T14484C15.
Se han encontrado muy pocos casos o familias que asocien el síndrome de Madelung con la mutación m8344A>G y el síndrome de MERRF y, hasta nuestro conocimiento, ninguna con solapamiento de la T14484C relacionada con LHON1, siendo esta la de mejor pronóstico. En este caso no pudimos realizar un estudio genético familiar, ya que no pertenecían a nuestra área sanitaria.
Como conclusión, queremos describir las manifestaciones oftalmológicas secundarias a este solapamiento de mutaciones mitocondriales.

