



La corea-acantocitosis (ChA) es una enfermedad autosómica recesiva que se produce por la aparición mutación localizada en el cromosoma 9q21 en el gen VPS13A que codifica una proteína llamada coreína. La coreína es una proteína de 3.000 aminoácidos relacionada con la formación del citoesqueleto celular, la exocitosis y la supervivencia celular1 sobre todo en neuronas estriatales2. El déficit de coreína puede activar mecanismos de autofagia y apoptosis. Se expresa en eritrocitos, neuronas, miocardiocitos, musculoesquelético y nervios periféricos. El cuadro típico se caracteriza por alteraciones psiquiátricas, cognitivas, tics y la característica distonía orofacial con mordeduras y automutilaciones de la lengua, los labios y los dedos. Los pacientes pueden asociar además la presencia de neuropatía periférica, aumento de las cifras de creatincinasa (CK), amiotrofia y crisis comiciales3.
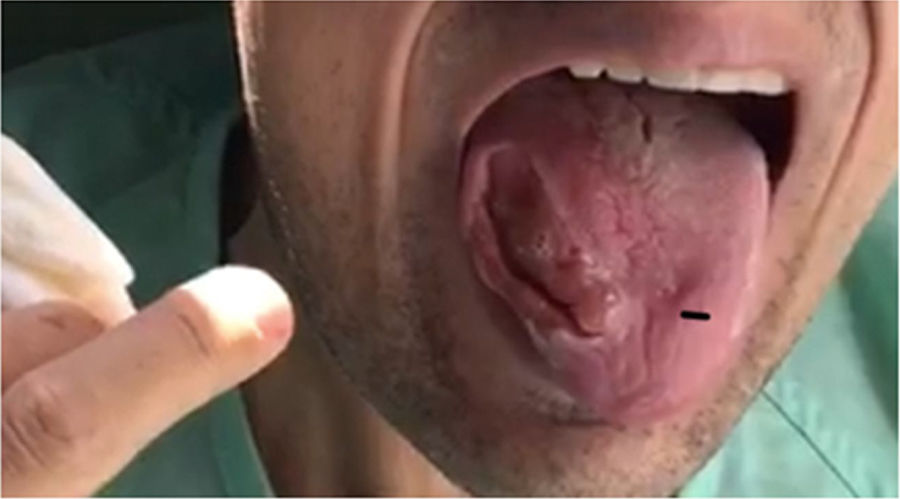
Varón de 28 años, segundo de una fratria de 3 hermanos, hijo de padres consanguíneos (primos carnales) sin antecedentes personales de interés salvo adenoidectomía en la infancia. Madre diagnosticada de epilepsia y demencia frontotemporal. En el año 2011 se realiza seguimiento por neurología y reumatología por detección de niveles elevados de CK y hormona adenocortitropa (ACTH) en analítica, sin filiarse la causa. En el 2013 sufre 2 crisis comiciales, instaurándose tratamiento con levetiracetam que decide abandonar. En el 2015 sufre mordedura lingual durante la ingesta, presentando desde entonces movimientos orofaciales distónicos e involuntarios con múltiples episodios de mordedura lingual con lesión severa de borde libre lingual derecho (fig. 1). Debido a la imposibilidad para una correcta ingesta presenta pérdida ponderal de 40kg en 2 años. Ingresa en psiquiatría en agosto del 2017 para estudio de cuadro obsesivo-compulsivo. Durante el ingreso se realiza un estudio por parte de medicina interna que incluye determinaciones analíticas de autoinmunidad, tóxicos, serologías y perfil general sin que se encuentren alteraciones salvo niveles elevados de CK. Estudio de electroencefalograma (EEG) y resonancia magnética (RM) cerebral con resultado normal. Debido a un nuevo episodio de mordedura lingual el paciente padece un cuadro séptico que obliga a realizar ingreso en la unidad de cuidados intensivos (UCI). Tras su estabilización es trasladado a neurología para continuar el estudio. Se le realiza un electroneurograma que revela presencia de polineuropatía axonal sensitivo-motora, así como un estudio de morfología de sangre periférica en el que se observan equinocitos. En planta sufre una nueva crisis comicial con desaturación por aspiración de las gasas que el paciente emplea para evitar las mordeduras constantes por lo que se instaura tratamiento con acetato de eslicarbacepina. Para el control conductual y de los movimientos distónicos orales se ensayan diferentes fármacos (clonazepam, haloperidol, tetrabenazina, pimozida, paliperidona, olanzapina, aripiprazol, naltrexona…) con respuesta escasa y tolerancia variable. Se realiza también infiltración de toxina botulínica en los músculos temporales, pterigoideos, maseteros y genioglosos para controlar mordedura lingual.

Dada la clínica compatible y la evolución claramente degenerativa de la sintomatología, se solicita panel genético de ChA descubriéndose presencia de mutación en homocigosis en gen VPS13A con transición de C por T (c.2347C>T) produciendo cambio de glutamina en posición 783 por codón de parada prematuro. Dicha mutación no había sido descrita con anterioridad, aunque en el contexto del paciente se supone patológica.
La ChA es una enfermedad de transmisión autosómica recesiva, poco prevalente4 y cuya sintomatología inicial puede ser confundida con un cuadro puramente psiquiátrico. Es importante sospecharla en pacientes que presentan una clínica compatible, sobre todo cuando existe historia de consanguineidad en la familia. El diagnóstico definitivo es genético ya que la presencia de acantocitos en sangre periférica no es constante5.

