






El objetivo es evaluar la eficacia y seguridad de los neuroestimuladores periféricos del ganglio esfenopalatino (GEP) para el tratamiento de la cefalea en racimos crónica refractaria al tratamiento.
DesarrolloRevisión sistemática de la literatura científica. Se identificaron estudios mediante una búsqueda en diferentes bases de datos. Las estrategias de búsqueda se realizaron hasta el 31 de octubre de 2016, incluyendo ensayos clínicos, revisiones sistemáticas o metaanálisis, informes de evaluación de tecnologías sanitarias y guías de práctica clínica que recogieran medidas de eficacia/efectividad o efectos adversos asociados al tratamiento. Se excluyeron estudios de cohortes, casos y controles, series de casos, revisiones narrativas, cartas al director, artículos de opinión, editoriales y estudios duplicados o desfasados por estudios posteriores de la misma institución. Respecto a la eficacia, los resultados son positivos tras la estimulación del GEP en relación con el alivio de dolor, el número de episodios, el uso de la medicación o la calidad de vida del paciente. En relación con la seguridad, hay un número importante de efectos adversos en los primeros 30 días de la intervención y en algunos pacientes fue necesaria la retirada del dispositivo. Los datos de seguimiento son a corto plazo y escasos.
ConclusionesLos resultados resultan prometedores a pesar de que la evidencia disponible es limitada. Consideramos fundamental continuar con la investigación sobre la seguridad y eficacia de los neuroestimuladores del GEP en la cefalea en racimos crónica. En aquellos casos en que pueda estar indicada la intervención, el tratamiento debería realizarse supervisado en un estudio de monitorización.
This study aimed to assess the safety and effectiveness of peripheral neurostimulation of the sphenopalatine ganglion (SPG) in the treatment of refractory chronic cluster headache.
DevelopmentVarious medical databases were used to perform a systematic review of the scientific literature. The search for articles continued until 31 October 2016, and included clinical trials, systematic reviews and/or meta-analyses, health technology assessment reports, and clinical practice guidelines that included measurements of efficiency/effectiveness or adverse effects associated with the treatment. The review excluded cohort studies, case-control studies, case series, literature reviews, letters to the editor, opinion pieces, editorials, and studies that had been duplicated or outdated by later publications from the same institution. Regarding effectiveness, we found that SPG stimulation had positive results for pain relief, attack frequency, medication use, and patients’ quality of life. In the results regarding safety, we found a significant number of adverse events in the first 30 days following the intervention. Removal of the device was necessary in some patients. Little follow-up data, and no long-term data, is available.
ConclusionsThese results are promising, despite the limited evidence available. We consider it essential for research to continue into the safety and efficacy of SPG stimulation for patients with refractory chronic cluster headache. In cases where this intervention may be indicated, treatment should be closely monitored.
La cefalea en racimos (CR) es una cefalea primaria, del grupo de las cefaleas trigémino-autonómicas, con aparición de episodios (racimos) de dolor periocular, unilaterales, de gran intensidad, junto con signos vegetativos ipsolaterales del lado doloroso, como ptosis, miosis, sudoración, lagrimeo, rinorrea, edema palpebral, o inquietud o agitación. Puede existir CR sin signos vegetativos ipsolaterales, siempre y cuando exista inquietud o agitación. Los ataques de dolor pueden durar entre 15 y 180 min y pueden repetirse entre 2 y 8 veces por día, con horario predominantemente nocturno. La CR puede presentarse en forma episódica —en la que se alternan los racimos con periodos libres de dolor que pueden prolongarse durante meses o años— y en forma crónica (CRC), con ausencia de fases de remisión de las crisis durante un año o más, o con remisiones que duran menos de un mes1.
Según los estudios, la incidencia de la CR oscila entre 2,5 y 9,8 casos/100.000 personas al año y la prevalencia entre 53 y 381 casos/100.000 personas. Parece ser más frecuente en hombres que en mujeres, con una proporción que varía en distintos estudios entre 7:1 y 3:12-7.
El tratamiento de la CR debe iniciarse suprimiendo los factores precipitantes del episodio, si los hubiere. El tratamiento farmacológico tiene un abordaje sintomático agudo de las crisis individuales (el fármaco de elección es el sumatriptán) y un abordaje profiláctico. La profilaxis puede hacerse mediante tratamientos preventivos de transición (con fármacos encaminados a cortar el episodio activo, como los esteroides, o mediante bloqueos anestésicos del nervio occipital) y de mantenimiento (con fármacos que consoliden la remisión y eviten una recaída precoz, como el verapamilo). El tratamiento preventivo debe mantenerse durante el racimo y suprimirse gradualmente tras un mes sin episodios2,8,9.
En pacientes sin respuesta al tratamiento (CRC refractarias), la opción alternativa es el tratamiento quirúrgico dirigido a la sección del trigémino, con más efectos secundarios y complicaciones. Recientemente, han aparecido alternativas como la neuroestimulación del ganglio esfenopalatino (GEP). El GEP es una colección ovoide de células parasimpáticas posganglionares sin función sensitiva. No obstante, presenta múltiples relaciones y conexiones con múltiples ramas faciales y trigeminales, y se cree que pueda estar implicado en la génesis y mantenimiento de dolores faciales atípicos y cefaleas unilaterales y, por tanto, puede ser considerado como diana terapéutica. Esta neuroestimulación del GEP puede realizarse mediante un dispositivo miniaturizado que se inserta por vía transoral utilizando una incisión bucogingival mínimamente invasiva. El sistema de neuroestimulación consta, además, de un controlador remoto que el paciente activa a demanda y de forma controlada cuando lo necesita y que permite al médico facilitar el ajuste de los parámetros que el paciente necesita8,9.
El objetivo de este trabajo es evaluar la eficacia y la seguridad de los neuroestimuladores periféricos del GEP para el tratamiento de la CRC refractaria al tratamiento.
DesarrolloSe abordó una revisión sistemática de la literatura científica mediante búsqueda en diferentes bases de datos (fig. 1). Se diseñaron estrategias de búsqueda para la identificación de estudios utilizando terminología libre y controlada, adaptando cada término al tesauro propio de cada base de datos (fig. 1). Se realizaron, igualmente, búsquedas en Internet y en los sitios web de las agencias de evaluación de tecnologías sanitarias (ETS) nacionales e internacionales, para localizar informes de ETS, si los hubiera, y una revisión manual de las referencias bibliográficas para localizar estudios no identificados con la búsqueda electrónica. La estrategia de búsqueda se realizó hasta el 31 de octubre de 2016 sin restricciones por tamaño de estudio ni idiomas.

Se incluyeron en la revisión ensayos clínicos aleatorizados, revisiones sistemáticas o metaanálisis, informes de ETS y guías de práctica clínica de pacientes con CRC refractaria al tratamiento tratados con neuroestimuladores periféricos del GEP y que recogieran medidas de eficacia/efectividad en términos de porcentaje de pacientes con reducción del dolor tras la estimulación, porcentaje de pacientes con desaparición completa del dolor tras la estimulación, reducción de la frecuencia de aparición de los episodios, calidad de vida medida con el Headache Impact Test (HIT), calidad de vida medida con el SF-36 o efectos adversos asociados al tratamiento.
Se excluyeron otros diseños de estudio, cartas al director, artículos de opinión, editoriales y estudios duplicados o desfasados por estudios posteriores de la misma institución.
La identificación, selección y revisión de los estudios la llevaron a cabo 2revisores de forma independiente. Se hizo una primera selección de estudios mediante la lectura de título y del abstract y, aquellos que en principio cumplían los criterios de inclusión, fueron revisados con la lectura del texto completo. Los desacuerdos se resolvieron por consenso junto con otro miembro del equipo investigador. Se elaboraron tablas con el detalle de los estudios incluidos y excluidos, justificando la causa de exclusión.
Se empleó el instrumento AMSTAR (Assessment of Multiple SysTemAtic Reviews) para evaluar la calidad de las revisiones sistemáticas y la herramienta de valoración del riesgo de sesgos de la Colaboración Cochrane para ensayos clínicos.
Los datos de los estudios seleccionados fueron extraídos por 2revisores de forma independiente. La recogida de datos incluía información sobre datos bibliográficos, características del estudio y de los pacientes, características de la intervención y medidas de resultado en relación con la eficacia. Igualmente, se recogió información sobre la seguridad del procedimiento.
Selección de estudiosSe identificaron 100 referencias (3 duplicadas). Tras la lectura del título y del resumen, 62 fueron excluidas por no cumplir los criterios de inclusión. Tras la lectura completa de las 35 publicaciones que cumplían los criterios de inclusión y exclusión, 29 fueron excluidas10-38 (fig. 2). De estas, 9 eran revisiones narrativas11,13,15,16,20-22,26,29, 8 revisiones sin datos17-19,24,25,27,28,31, 8 abstracts14,32-38, 3 no eran de CRC10,12,30 y una publicación no era de neuroestimuladores periféricos23. Finalmente, fueron seleccionados 6 estudios: un informe de ETS de la agencia austriaca realizado con metodología de revisión sistemática39, un informe NICE40, un ensayo clínico (ensayo Pathway CH-1) registrado en clinicaltrials.org (NCT 01255813)41, 2 artículos42,43 con datos de un estudio de seguimiento a largo plazo (24 meses) de los pacientes del ensayo Pathway CH-1 registrado en clinicaltrials.org (NCT 01616511) y un registro poscomercialización44 registrado en clinicaltrials.org (NCT 01677026) que incluía, entre otros, a los pacientes del ensayo Pathway CH-1, por lo que se decidió incluirlo en este estudio. Para la elaboración de esta revisión se extrajeron los datos de los estudios originales, todos financiados por Autonomic Technologies, Inc (ATI), empresa fabricante del neuroestimulador.
Resultados de eficacia
Tanto el informe NICE40 como el informe de ETS austriaco39 se basan en los datos que reportaba el estudio Pathway CH-141 realizado con 32 pacientes que recibieron de forma aleatorizada estimulación completa del GEP, estimulación subumbral o estimulación simulada durante cada episodio de CR. La estimulación del GEP se realizó mediante el dispositivo ATI Neurostimulation System, también denominado ATI Neuroestimulator y PulsanteTM SPG Microstimulator45.
El ensayo Pathway CH-141 consideró como CR refractaria a aquella con un mínimo de 4 episodios de dolor de cabeza graves por semana que afectan la calidad de vida y que no han respondido a por lo menos 3 tratamientos preventivos consecutivos con la dosis máxima tolerable. El estudio consta de 5 fases: periodo basal antes del implante del neuroestimulador, periodo de estabilización postimplante, periodo de valoración de la terapia para el ajuste de electrodos y parámetros de estimulación, periodo experimental en el que se produce la aleatorización a estimulación completa, subumbral o simulada y periodo abierto en el que todos los pacientes se tratan con estimulación completa.
Un total de 28 pacientes terminaron el periodo experimental. El ensayo encontró los siguientes resultados (tabla 1): la reducción del dolor a los 15 min tras la neuroestimulación fue del 67% de los episodios tratados con estimulación completa frente al 7% de los tratados con estimulación subumbral y el 7% de los tratados con estimulación simulada (p<0,0001). La resolución completa del dolor a los 15 min tras la neuroestimulación fue del 34% de los episodios tratados con estimulación completa, frente al 2% de los tratados con estimulación subumbral y al 2% de los tratados con estimulación simulada (p<0,0001). Se observó una disminución del dolor a los 30, 60 y 90 min tras la neuroestimulación en el 56, 61 y 60%, respectivamente, de los episodios de CR tratados con estimulación completa frente al 8, 12 y 13% de los tratados con estimulación simulada (p<0,0001). La frecuencia media de episodios disminuyó de 17,4 a 12,5 episodios por semana para los 28 pacientes que completaron el periodo experimental (p=0,005). Un 68% de los pacientes respondieron al tratamiento de neuroestimulación, de los cuales el 25% consiguió alivio del dolor en ≥50% de los episodios tratados, un 36% consiguió una reducción ≥50% en la frecuencia de los episodios y un 7% consiguió ambas cosas. El uso de medicación durante la fase aguda se produjo en el 31% de los episodios tratados con estimulación completa frente al 78% de los tratados con estimulación subumbral y el 77% con estimulación simulada (p=0,0001). El HIT mejoró en 6,8±10,2 puntos entre el periodo base y el experimental (p=0,002). La calidad de vida evaluada con la SF-36 mejoró en el 75% de pacientes; la función física en el 21%, la función mental en el 29% y un 25% de los pacientes mejoraron en ambas funciones.
Resultados de eficacia recogidos en el ensayo Pathway CH1 y en el seguimiento a 24 meses
| Ensayo Pathway CH-1 | Estudio de seguimiento prolongado del Pathway CH-1 sin comparador (24 meses) | ||
|---|---|---|---|
| Schoenen et al., 2013 | Barloese et al., 2016 (Da resultados de casoscon remisión de los episodios) | Jurgens et al., 2016 | |
| N.° de pacientes | Periodo base: 32Periodo experimental: 28 | n=10 (el estudio sigue los pacientes con remisión de los episodios de al menos un mes, n=10 (30%) | n=33. Datos completos de seguimiento de 31 pacientes |
| Alivio dolor a 15min | 127/190 (67%) EC vs. 14/184 (7%) ESU (p < 0,0001)127/190 (67%) EC vs. 15/192 (7%) ES (p < 0,0001)14/184 (7%) ESU vs. 7,4% ES (p=0,96) | --- | --- |
| Libre de dolor a 15min | 65/190 (34%) EC vs. 3/184 (2%) ESU (p < 0,0001)65/190 (34%) EC vs. 3/192 (2%) ES (p < 0,0001)3/184 (2%) vs. 3/192 (2%) ES (p < 0,97) | --- | Libre de dolor en el seguimiento: 50% de los episodios tratados |
| Alivio del dolor a 30min (% episodios) | 56% EC vs. 8% ES (p < 0,0001) | --- | --- |
| Alivio del dolor a 60min (% episodios) | 61% EC vs. 12% ES (p < 0,0001) | --- | --- |
| Alivio del dolor a 90min (% episodios) | 60% EC vs. 13% ES (p < 0,0001) | --- | --- |
| Uso de medicación (% de episodios) | 31% EC vs. 78% ESU (p < 0,0001)31% EC vs. 77% ES (p < 0,0001)44% ES vs. 78% ESU (p < 0,68) | Mejoría en 4 pacientes (40%) tras la remisión de los episodios y en 6 (60%) pacientes a los 24 meses | 21/33 (64%) mejoraron clínicamente respecto al uso de medicación a los 24 meses de seguimiento |
| Frecuencia episodios/ semana (media) | Periodo base: 17,4 (rango: 4-70) vs. periodo experimental: 12,5 (rango: 0-96) (p=0,005) | Periodo base: 17,3±13,3Al iniciar la estimulación: 16,4±16,8 (p=0,7090) | No cambió: periodo base: 17±3; a los 24 meses: 17±15 |
| Tipo de pacientes | 19/28 pacientes (68%) respondieron al tratamiento:- Respondedor agudo: 7 (25%)- Respondedor frecuente: 10 (36%)- Respondedor terapéutico: 2 (7%) | --- | - Respondedor agudo: 15/33 (45%). El 78% de estos, terapia efectiva- Respondedor frecuente: 11/31 (35%)- Respondedor terapéutico: 20/33 (61%) |
| Escala HIT-6 | Diferencia entre periodo experimental y base de −6,8±10,2 unidades (p=0,002); 18/28 pacientes (64%) mejoraron más que la diferencia media en esta escala: −2,3 unidades | HTI-6 varió de 67,7±6,0 (rango: 58-76) en periodo base a 55,2±11,4 tras la remisión (rango: 40-73) (p=0,0118) y a 60,0±8,5 a los 24 meses (p=0,1997) | Reducción en 4,8 unidades a los 24 meses en relación con el periodo base en 29 pacientes (p=0,0048) (4 pacientes no completaron el seguimiento) |
| Escala SF-36v2 | Mejoró en 21/28 pacientes (75%):- 6 (21%) en la función física- 8 (29%) en la función mental- 7 (25%) en ambas | --- | --- |
| Satisfacción personal | --- | En todos los pacientes (100%) | El 69% de los pacientes que responden (18/26) encontraron el tratamiento útil |
| Episodios contralaterales | --- | --- | 11/33 pacientes (33%) |
EC: estimulación completa; ES: estimulación simulada; ESU: estimulación subumbral.
Dos artículos, con 33 pacientes, publicaron el estudio de seguimiento a 24 meses de los pacientes incluidos en el ensayo Pathway CH-142,43. El estudio de Barloese et al.42 siguió a 10 pacientes con remisión de los episodios al menos durante un mes tras la estimulación del GEP y mostró los siguientes resultados (tabla 1): un 30% de los pacientes experimentaron uno o más periodos de remisión de al menos un mes; el primer periodo de remisión comenzó 134±86 días después del inicio de la estimulación (rango: 21-272 días); la media del periodo de remisión más largo fue de 149±97 días (rango: 62-322); el 60% de los pacientes con remisión consiguió disminuir el uso de medicamentos preventivos a los 24 meses. El HIT tras la remisión mejoró con respecto a las cifras basales: 67,7±6,0 frente a 55,2±11,4 tras la remisión (p=0,0118) y frente a 60,0±8,5 a los 24 meses (p=0,1997). A los 24 meses tras la inserción, la mejoría se mantuvo y las medidas de satisfacción del paciente fueron positivas en el 100% de los pacientes con remisión.
El estudio de Jurgens et al.43 recogió datos de seguimiento de los 33 pacientes tras 24 meses (tabla 2): el 50% de los episodios tratados alcanzaron la resolución completa de dolor. Un 64% de los pacientes mantuvo su respuesta terapéutica a los 24 meses. La frecuencia media de episodios, en los 33 pacientes, no varió en el periodo base (17±3) frente al de seguimiento (17±15). A los 24 meses, el 45% tuvo una respuesta aguda a la estimulación y el 35% consiguió una respuesta frecuente. En total, el 61% fueron respondedores terapéuticos. A los 24 meses, el HIT se redujo en 4,8 unidades respecto a las cifras basales en los 29 pacientes que completaron esta escala (p=0,0048). El 69% de los pacientes manifestó que el tratamiento fue útil y, durante los 24 meses, el 33% de los pacientes informó de episodios de CR en el lado opuesto a la inserción del neuroestimulador.
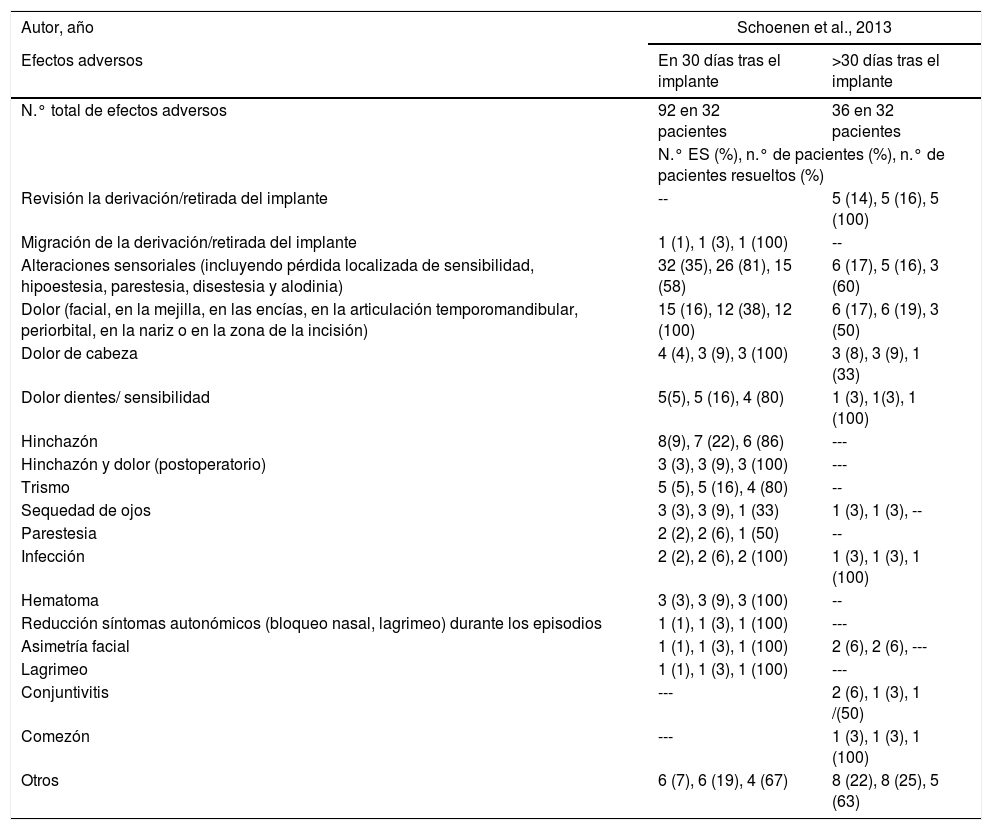
Efectos adversos, desde la implantación del dispositivo al final del periodo experimental, recogidos en el ensayo Pathway CH-1
| Autor, año | Schoenen et al., 2013 | |
|---|---|---|
| Efectos adversos | En 30 días tras el implante | >30 días tras el implante |
| N.° total de efectos adversos | 92 en 32 pacientes | 36 en 32 pacientes |
| N.° ES (%), n.° de pacientes (%), n.° de pacientes resueltos (%) | ||
| Revisión la derivación/retirada del implante | -- | 5 (14), 5 (16), 5 (100) |
| Migración de la derivación/retirada del implante | 1 (1), 1 (3), 1 (100) | -- |
| Alteraciones sensoriales (incluyendo pérdida localizada de sensibilidad, hipoestesia, parestesia, disestesia y alodinia) | 32 (35), 26 (81), 15 (58) | 6 (17), 5 (16), 3 (60) |
| Dolor (facial, en la mejilla, en las encías, en la articulación temporomandibular, periorbital, en la nariz o en la zona de la incisión) | 15 (16), 12 (38), 12 (100) | 6 (17), 6 (19), 3 (50) |
| Dolor de cabeza | 4 (4), 3 (9), 3 (100) | 3 (8), 3 (9), 1 (33) |
| Dolor dientes/ sensibilidad | 5(5), 5 (16), 4 (80) | 1 (3), 1(3), 1 (100) |
| Hinchazón | 8(9), 7 (22), 6 (86) | --- |
| Hinchazón y dolor (postoperatorio) | 3 (3), 3 (9), 3 (100) | --- |
| Trismo | 5 (5), 5 (16), 4 (80) | -- |
| Sequedad de ojos | 3 (3), 3 (9), 1 (33) | 1 (3), 1 (3), -- |
| Parestesia | 2 (2), 2 (6), 1 (50) | -- |
| Infección | 2 (2), 2 (6), 2 (100) | 1 (3), 1 (3), 1 (100) |
| Hematoma | 3 (3), 3 (9), 3 (100) | -- |
| Reducción síntomas autonómicos (bloqueo nasal, lagrimeo) durante los episodios | 1 (1), 1 (3), 1 (100) | --- |
| Asimetría facial | 1 (1), 1 (3), 1 (100) | 2 (6), 2 (6), --- |
| Lagrimeo | 1 (1), 1 (3), 1 (100) | --- |
| Conjuntivitis | --- | 2 (6), 1 (3), 1 /(50) |
| Comezón | --- | 1 (3), 1 (3), 1 (100) |
| Otros | 6 (7), 6 (19), 4 (67) | 8 (22), 8 (25), 5 (63) |
El ensayo Pathway CH-141 informó de 128 efectos adversos en 32 pacientes (tabla 2). En un paciente se retiró el dispositivo en los primeros 30 días tras la intervención. La revisión de la derivación del neuroestimulador o retirada del dispositivo fue necesaria en el 16% de los pacientes, entre 30 días y un año después del procedimiento. Hubo alteraciones sensoriales (incluyendo pérdida localizada de sensibilidad, hipoestesia, parestesia, disestesia y alodinia) en el 81% de los pacientes en los 30 días posteriores a la implantación del dispositivo, con resolución de los síntomas en el 58% de estos pacientes. Se notificaron alteraciones sensoriales en un 16% de los pacientes entre 30 días y un año después del procedimiento, resolviéndose los síntomas en el 60%. En los primeros 30 días de la implantación, el 38% refirió dolor (facial, en la mejilla, en las encías, en la articulación temporomandibular, periorbital, en la nariz o en la zona de la incisión). La gravedad del dolor no se describió y los síntomas se resolvieron en todos los pacientes. Un 19% refirió dolor entre 30 días y un año después del procedimiento, síntomas que se resolvieron en el 50% de los casos. El 9% de los pacientes presentó cefaleas que no eran CR en los primeros 30 días posteriores a la implantación del dispositivo, resolviéndose los síntomas en todos los casos. Igualmente, un 9% presentó dolores de cabeza, que no eran CR entre 30 días y un año después del procedimiento, resolviéndose los síntomas en el 33% de casos. Un 22% de los pacientes presentó inflamación específica en los primeros 30 días tras la implantación del dispositivo, resolviéndose en un 86% de los casos.
Un 16% de los pacientes presentó trismo en los primeros 30 días tras la implantación del dispositivo, que se resolvió en el 80% de casos. Un 9% presentó sequedad de ojos en los primeros 30 días tras la implantación, resuelto en el 33% de los pacientes. Un 3% presentó sequedad de ojos entre 30 días y un año después del procedimiento, sin resolución y un 6% presentaron parestesia leve de los músculos alrededor del pliegue nasolabial en los primeros 30 días tras la implantación, resolviéndose en la mitad de los pacientes. Dos pacientes (6%) presentaron infección en los primeros 30 días tras la implantación, que se resolvieron tras tratamiento con antibióticos.
Se recogieron otros efectos adversos en los primeros 30 días tras la implantación del dispositivo como hematomas, reducción de síntomas autonómicos durante los episodios y asimetría facial, resolviéndose en la mayoría de los pacientes. Por otra parte, entre 30 días y un año después del procedimiento, se recogieron otros efectos adversos como conjuntivitis o comezón, que desaparecieron en la mayoría de los pacientes (tabla 2).
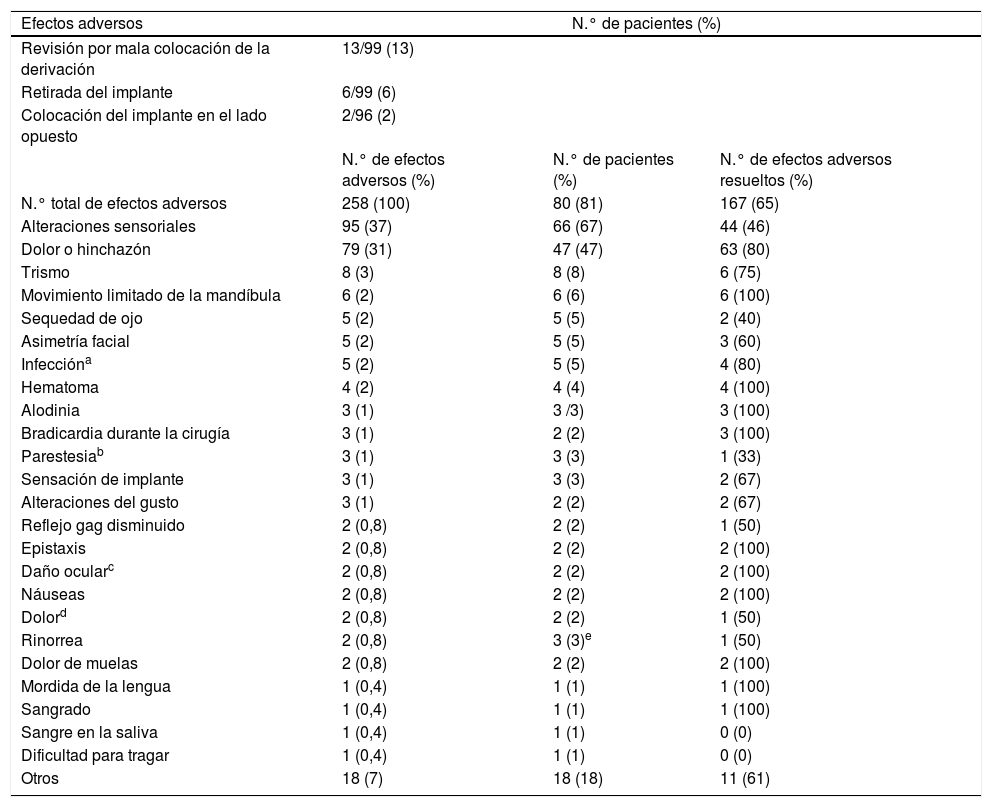
El registro de 99 pacientes44 incluyó, además de los 32 del ensayo clínico Pathway CH-141, 11 pacientes con acceso abierto al ensayo Pathway CH-1, y los pacientes del registro Pathway R-1 (total n=99). Esta publicación recogió principalmente datos de efectos adversos durante el periodo perioperatorio (30 días tras la intervención) en los pacientes del registro (tabla 3). Además, el estudio informó de la realización de distintos procedimientos de seguimiento: colocación de un segundo neuroestimulador en el lado opuesto de la cara (n=2), ajuste de la localización de la derivación del neuroestimulador dentro de la fosa pterigopalatina para intentar dirigir mejor el GEP (n=13) y retirada del neuroestimulador por diferentes causas (n=6).
Efectos adversos perioperatorios (30 días después del implante) que recoge el registro de pacientes publicado por Assaf et al.. (n=99)
| Efectos adversos | N.° de pacientes (%) | ||
| Revisión por mala colocación de la derivación | 13/99 (13) | ||
| Retirada del implante | 6/99 (6) | ||
| Colocación del implante en el lado opuesto | 2/96 (2) | ||
| N.° de efectos adversos (%) | N.° de pacientes (%) | N.° de efectos adversos resueltos (%) | |
| N.° total de efectos adversos | 258 (100) | 80 (81) | 167 (65) |
| Alteraciones sensoriales | 95 (37) | 66 (67) | 44 (46) |
| Dolor o hinchazón | 79 (31) | 47 (47) | 63 (80) |
| Trismo | 8 (3) | 8 (8) | 6 (75) |
| Movimiento limitado de la mandíbula | 6 (2) | 6 (6) | 6 (100) |
| Sequedad de ojo | 5 (2) | 5 (5) | 2 (40) |
| Asimetría facial | 5 (2) | 5 (5) | 3 (60) |
| Infeccióna | 5 (2) | 5 (5) | 4 (80) |
| Hematoma | 4 (2) | 4 (4) | 4 (100) |
| Alodinia | 3 (1) | 3 /3) | 3 (100) |
| Bradicardia durante la cirugía | 3 (1) | 2 (2) | 3 (100) |
| Parestesiab | 3 (1) | 3 (3) | 1 (33) |
| Sensación de implante | 3 (1) | 3 (3) | 2 (67) |
| Alteraciones del gusto | 3 (1) | 2 (2) | 2 (67) |
| Reflejo gag disminuido | 2 (0,8) | 2 (2) | 1 (50) |
| Epistaxis | 2 (0,8) | 2 (2) | 2 (100) |
| Daño ocularc | 2 (0,8) | 2 (2) | 2 (100) |
| Náuseas | 2 (0,8) | 2 (2) | 2 (100) |
| Dolord | 2 (0,8) | 2 (2) | 1 (50) |
| Rinorrea | 2 (0,8) | 3 (3)e | 1 (50) |
| Dolor de muelas | 2 (0,8) | 2 (2) | 2 (100) |
| Mordida de la lengua | 1 (0,4) | 1 (1) | 1 (100) |
| Sangrado | 1 (0,4) | 1 (1) | 1 (100) |
| Sangre en la saliva | 1 (0,4) | 1 (1) | 0 (0) |
| Dificultad para tragar | 1 (0,4) | 1 (1) | 0 (0) |
| Otros | 18 (7) | 18 (18) | 11 (61) |
Infección: nariz/sinusitis; sitio de la incisión: resuelto con antibióticos; boca: resuelto con antibióticos; sitio de la incisión: resuelto con retirada, resueltos con antibióticos; apertura de la cicatriz: resuelto con antibióticos.
Las principales secuelas quirúrgicas fueron las alteraciones sensoriales (67% de pacientes) con un total de 95 alteraciones sensoriales diferentes, 44 de ellas resueltas en una media de 104 días (rango: 12-313). El dolor/hinchazón fue referido por el 47% de los pacientes con un total de 79 eventos, de los cuales 63 se resolvieron en una media de 68 días (rango: 0-312). El 92% de los eventos fueron calificados por el paciente como leves o moderados en su impacto en las actividades diarias. Finalmente, 29 de los 32 del registro completaron un cuestionario de autoevaluación a los 18 meses del implante: el 86% consideró que los efectos de la cirugía fueron tolerables y el 90% indicó que tomaría de nuevo la misma decisión para tratar su CR.
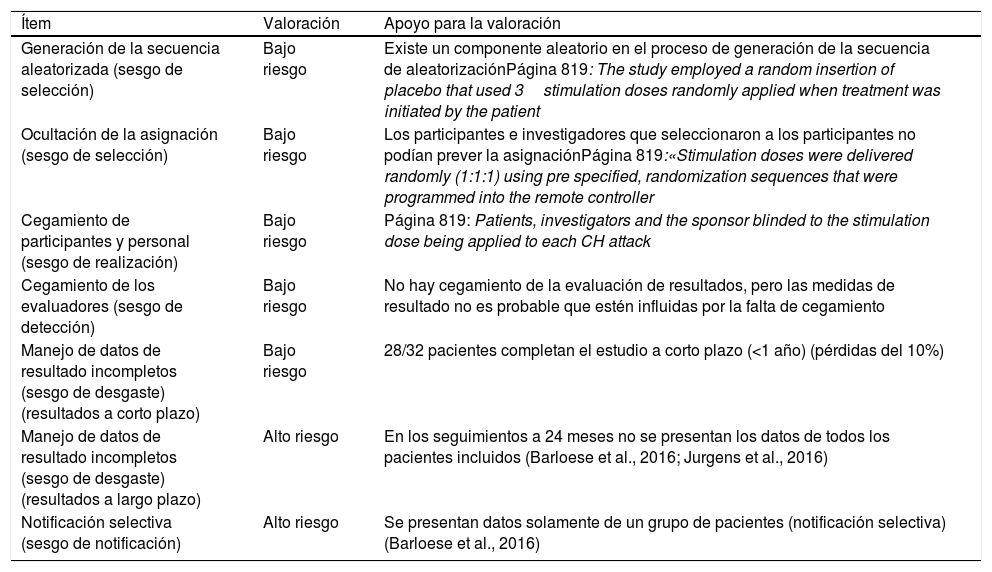
Resultados de la evaluación de sesgos y calidad de los estudiosEn la valoración de sesgos de la Colaboración Cochrane, el ensayo Pathway CH-141 presentaba bajo riesgo de sesgos en algunos aspectos (tabla 4). No obstante, los estudios que informaban del seguimiento a largo plazo de los pacientes del ensayo presentaban alto riesgo de sesgo para los datos a largo plazo y la notificación selectiva, ya que se recogían datos solamente de un grupo seleccionado de pacientes.
Valoración de sesgos de la Colaboración Cochrane. Resultados a corto plazo (Schoenen et al., 2013) y seguimiento a 24 meses (Barloese et al., 2016; Jurgens et al., 2016)
| Ítem | Valoración | Apoyo para la valoración |
| Generación de la secuencia aleatorizada (sesgo de selección) | Bajo riesgo | Existe un componente aleatorio en el proceso de generación de la secuencia de aleatorizaciónPágina 819: The study employed a random insertion of placebo that used 3stimulation doses randomly applied when treatment was initiated by the patient |
| Ocultación de la asignación (sesgo de selección) | Bajo riesgo | Los participantes e investigadores que seleccionaron a los participantes no podían prever la asignaciónPágina 819:«Stimulation doses were delivered randomly (1:1:1) using pre specified, randomization sequences that were programmed into the remote controller |
| Cegamiento de participantes y personal (sesgo de realización) | Bajo riesgo | Página 819: Patients, investigators and the sponsor blinded to the stimulation dose being applied to each CH attack |
| Cegamiento de los evaluadores (sesgo de detección) | Bajo riesgo | No hay cegamiento de la evaluación de resultados, pero las medidas de resultado no es probable que estén influidas por la falta de cegamiento |
| Manejo de datos de resultado incompletos (sesgo de desgaste) (resultados a corto plazo) | Bajo riesgo | 28/32 pacientes completan el estudio a corto plazo (<1 año) (pérdidas del 10%) |
| Manejo de datos de resultado incompletos (sesgo de desgaste) (resultados a largo plazo) | Alto riesgo | En los seguimientos a 24 meses no se presentan los datos de todos los pacientes incluidos (Barloese et al., 2016; Jurgens et al., 2016) |
| Notificación selectiva (sesgo de notificación) | Alto riesgo | Se presentan datos solamente de un grupo de pacientes (notificación selectiva) (Barloese et al., 2016) |
En cuanto al informe de ETS austriaco39, consideramos que es de buena calidad, con una puntuación de 8 sobre 10 en el instrumento AMSTAR. Presentaba un diseño «a priori»; al menos 2personas seleccionaron los estudios y extrajeron la información; la estrategia de búsqueda fue amplia y rigurosa; el tipo y estado de la publicación no se usó como criterio de inclusión; se explicitaron las características de los estudios; se evaluó y documentó la calidad de los estudios; se utilizó adecuadamente esta calidad en la formulación de conclusiones y se declararon los conflictos de interés, aunque no se adjuntó una lista de los estudios incluidos y excluidos.
DiscusiónEl presente estudio evalúa los neuroestimuladores periféricos del GEP como nueva alternativa terapéutica para los pacientes con CRC refractaria al tratamiento. Aunque el número de personas afectadas por el proceso es reducido, esta tecnología es importante y supone un gran avance para los pacientes con CRC refractaria a los tratamientos farmacológicos y a los que solo les queda la opción quirúrgica, con complicaciones graves en algunos casos8.
Si bien los estudios de cohortes o casos y controles pudieran aportar datos complementarios de interés, el objetivo de este estudio fue evaluar la eficacia y seguridad de los neuroestimuladores del GEP y, por ello, se seleccionaron ensayos clínicos, revisiones sistemáticas o metaanálisis por ser los diseños metodológicos que aportan información de mayor calidad. En cuanto a la seguridad, sí se incluyó un registro de pacientes con datos acerca de posibles efectos adversos del uso de neuroestimuladores del GEP.
Hemos encontrado estudios que analizan la efectividad y seguridad de los neuroestimuladores periféricos del GEP en la CRC. Sin embargo, observamos que la información recogida en estos estudios proviene esencialmente de pacientes incluidos en un único ensayo clínico. Dicho ensayo, de un año de duración, incluye a 32 pacientes diagnosticados con CRC refractaria al tratamiento. Sin embargo, observamos que el estudio experimental con aleatorización y uso de un comparador, es solo de 3 a 8 semanas y lo completan 28 pacientes41. Datos de seguimiento a 24 meses (que informa de 33 pacientes, uno más que el ensayo Pathway CH-1), recogidos en otras publicaciones, informan solo sobre algunos indicadores de eficacia42,43.
El registro poscomercialización con 99 casos incluye los 32 del ensayo clínico44 y, por ello, decidimos su inclusión en esta revisión sistemática. Recoge esencialmente los efectos adversos en los primeros 30 días tras la intervención. Por tanto, observamos que la información sobre eficacia y seguridad del neuroestimulador del GEP en la CRC es limitada.
Al valorar la factibilidad del neuroestimulador del GEP, vemos que constituye una alternativa terapéutica, fácil de usar por el paciente y factible45; sin embargo, los datos del ensayo clínico muestran que en 6 pacientes fue necesaria la revisión de la derivación del neuroestimulador y la retirada del dispositivo durante el primer año tras la implantación41. El registro postintervención informa de la necesidad de revisión de la derivación en el 13% de los pacientes por colocación inapropiada y recoge la retirada del dispositivo en el 6% de los casos en los primeros días de la intervención44. Las causas descritas para la retirada del dispositivo incluyen dolor por afectación del nervio maxilar, desplazamiento, colocación inadecuada del electrodo o infección de la incisión quirúrgica40. Todo ello invita a la cautela y a la necesidad de supervisar e investigar el procedimiento40.
Respecto a la eficacia del dispositivo, observamos que el ensayo clínico encuentra resultados estadísticamente significativos tras la estimulación del GEP en relación con el alivio o ausencia de dolor, la disminución del número de episodios semanales o el uso de la medicación, comparando la estimulación completa con la subumbral o la simulada. También encuentra resultados significativos a favor de la intervención en la escala HIT-6 y la SF-36. No obstante, aunque los resultados resultan prometedores, es preciso interpretarlos con cautela. Los datos proceden de un único ensayo clínico, de corta duración, con un número reducido de pacientes, con algunas pérdidas y en el que el procedimiento de cegamiento puede ser incierto, pues los pacientes pueden ser conscientes de la dosis de estimulación recibida41. Por otra parte, en la historia natural de la CRC puede haber periodos de remisión espontánea, lo que debería ser tenido en cuenta42. Los datos de seguimiento a 24 meses muestran que algunos pacientes presentan resultados favorables en este periodo respecto a la situación basal. Sin embargo, el número de casos es limitado y los datos de seguimiento son escasos42,43. Por otra parte, se han encontrado casos de CR contralateral durante el seguimiento de los pacientes sin causa aparente, por lo que se considera necesario continuar la investigación sobre los efectos del neuroestimulador del GEP43.
En relación con la seguridad, encontramos que los estudios recogen un número importante de efectos adversos en los primeros 30 días de la implantación del neuroestimulador del GEP. De hecho, el ensayo clínico encuentra que hasta un 81% de los pacientes presenta alteraciones sensoriales, y un alto porcentaje de pacientes refiere dolor o hinchazón en dicho periodo. No obstante, la mayoría de los episodios se pudieron resolver con éxito41. El número de complicaciones disminuye a partir de los 30 días de la intervención, aunque aún un número importante de pacientes presenta efectos adversos en este periodo. El registro postintervención44 recoge que más de la mitad de los pacientes presenta algún efecto adverso, aunque la mayor parte de ellos fueron considerados por los pacientes como leves o moderados. Algunos autores consideran que, aunque el número de efectos adversos es alto, las complicaciones de la intervención podrían ser comparables a las de otros procedimientos de la cavidad oral como extracciones dentales, cirugía de senos o implantes dentales44. En cuanto a la seguridad del procedimiento a largo plazo, los estudios que recogen datos de seguimiento a 24 meses no incluyen información sobre efectos adversos en dicho periodo. Únicamente Jurgens et al. hacen referencia a que los efectos adversos encontrados fueron leves o moderados y se resolvieron en el plazo de 2-3 meses43.
A la hora de valorar los resultados de eficacia y seguridad de los neuroestimuladores del GEP es preciso tener en cuenta las limitaciones de los estudios. El informe NICE40 considera que el ensayo clínico debería describir en detalle los criterios de selección de los pacientes, la selección debería ser realizada por un equipo especialista en tratamiento del dolor y se debería informar en detalle de la intensidad y duración de la estimulación, del uso de medicación, la calidad de vida y de los efectos sobre los síntomas40. Por otra parte, los estudios incluidos en nuestra revisión no son independientes, y no hemos encontrado publicaciones que estudien la eficacia del neuroestimulador en relación con otros tratamientos médicos o quirúrgicos, lo que ayudaría a conocer mejor la eficacia del procedimiento. Aunque algunos autores hablan de resultados del neuroestimulador del GEP a largo plazo42, el periodo de seguimiento máximo recogido en la literatura es de 24 meses, por lo que consideramos que no se dispone de suficiente información sobre resultados del procedimiento a largo plazo.
Los informes de ETS que evalúan la eficacia y seguridad de los neuroestimuladores del GEP39,40 resaltan la frecuencia de efectos adversos e incluso la necesidad en ocasiones de retirar el dispositivo y concluyen que el procedimiento no debería prestarse en las condiciones clínicas habituales, sino dentro de un programa de investigación40. El informe austriaco recomienda, con las pruebas disponibles, la no inclusión del procedimiento en el catálogo de prestaciones sanitarias del sistema sanitario del país39. En nuestra opinión, los datos de seguimiento de los pacientes a 24 meses analizados en nuestro estudio no aportan suficiente información para poder contradecir las conclusiones.
En relación con la satisfacción del paciente, observamos que la mayoría de los pacientes encuestados se mostraron satisfechos con la intervención recibida42 y encontraron el tratamiento útil43. El 90% de los pacientes del ensayo clínico afirmó, en un cuestionario de autoevaluación realizado a los 18 meses de la intervención, que volvería a tomar la misma decisión terapéutica para tratar su CRC44. En este sentido, creemos importante señalar que el neuroestimulador periférico del GEP puede constituir la única alternativa terapéutica a una necesidad no cubierta, ya que determinados pacientes no responden a los tratamientos habituales46. No obstante, la implicación del paciente y el consentimiento informado escrito se considera un requisito fundamental ante la elección del tratamiento40.
ConclusionesLa información de los estudios que evalúan el tratamiento de los neuroestimuladores del GEP para la CRC refractaria proviene esencialmente de un único ensayo clínico de corta duración y con pocos pacientes. Aunque los estudios incluidos en esta revisión presentan limitaciones, no son independientes y no evalúan la eficacia del neuroestimulador en comparación con otros tratamientos médicos o quirúrgicos, es importante señalar que la neuroestimulación periférica del GEP puede constituir la única alternativa terapéutica en pacientes con CRC que no responden al tratamiento.
En relación con la eficacia, encontramos resultados positivos tras la estimulación del GEP en cuanto al alivio del dolor, número de episodios, uso de medicación, satisfacción con el tratamiento recibido o calidad de vida. A los 24 meses de la intervención, algunos pacientes presentan resultados favorables respecto a la situación basal, aunque los datos de seguimiento son escasos.
En relación con la seguridad, encontramos un número importante de efectos adversos en los primeros 30 días de la intervención. En ocasiones, fue necesaria la retirada del dispositivo. No existen datos de seguridad del procedimiento a largo plazo.
Los resultados de eficacia y seguridad resultan prometedores, aunque la evidencia disponible es limitada. Consideramos fundamental continuar con la investigación sobre la seguridad y eficacia de los neuroestimuladores del GEP en pacientes con CRC refractaria al tratamiento. En aquellos casos en los que la intervención pueda estar indicada, esta debería realizarse supervisada, bajo un protocolo de investigación, prestando especial atención al consentimiento informado escrito del paciente.
Conflicto de interesesLos autores declaran no tener conflictos de intereses.

