



La falta de criterios homogéneos aceptados para la definición de algunas de las patologías desmielinizantes dificulta la caracterización diagnóstica limitando la reproducibilidad de los resultados y las recomendaciones terapéuticas. Especialmente controvertidas son las formas de encefalomielitis recurrentes (EAD-RR) y otras formas infrecuentes de neuromielitis óptica (NMO).
ObjetivoDescribimos la evolución clínico-radiológica de un caso de EAD-RR del adulto versus NMO, seguida durante 9 años.
Paciente y métodosLa paciente debutó con síntomas severos de rombencefalomielitis y la resonancia magnética (RM) craneal y medular mostraron lesiones extensas, con captación de gadolinio en el tronco encefálico y de la médula, acorde con los síntomas clínicos de la paciente. Se excluyó etiología infecciosa, el índice IgG fue normal y fueron negativos los anticuerpos para NMO. Tras tratamiento con corticoides por vía intravenosa y plasmaféresis la recuperación del episodio fue excelente. Durante el seguimiento ha presentado 7 recurrencias, preferentemente medulares, con buena recuperación, que reproducen con severidad variable los mismos síntomas. Desde el inicio ha recibido tratamiento inmunosupresor.
ConclusionesNuestro caso comparte características clínicas con EAD-RR y NMO e ilustra que, pese a los criterios vigentes, la caracterización diagnóstica de estas entidades no es fácil.
The lack of accepted homogeneous criteria for the definition of some demyelinating diseases makes diagnostic characterization difficult and limits data interpretation and therapeutic recommendations. Recurrent encephalomyelitis (ADE-R) along with borderline cases of neuromyelitis optica (NMO) are especially controversial.
ObjectiveTo describe the clinical and radiological evolution of an adult-onset ADE-R versus NMO case throughout 9 years of follow-up.
Patient and methodsOur patient presented with severe symptoms of rhombencephalomyelitis and the cranial and spinal magnetic resonance imaging (MRI) showed large lesions, with gadolinium enhancement in brainstem and spinal cord, correlating with the clinical picture. Infectious aetiology was excluded, IgG index was normal and NMO antibodies were negative. After treatment with intravenous corticosteroids and plasmapheresis, there was excellent recovery in the acute phase. During follow-up, seven relapses have occurred, mainly in the spinal cord, with good recovery and the same symptomatology, albeit with different severity. Immunosuppressive treatment was introduced since the beginning.
ConclusionsOur case shares common features of both ADE-R and NMO, illustrating that diagnostic characterization is not easy in spite of current criteria.
La encefalomielitis aguda diseminada (EAD) idiopática es una enfermedad inflamatoria desmielinizante que afecta fundamentalmente a niños, habitualmente precedida por infecciones víricas o vacunas, con latencia de 2 a 30 días y mayor incidencia en invierno1–4. Se deben excluir causas infecciosas5–7 y neoplásicas8. La hipótesis etiopatogénica es una respuesta inmunitaria inadecuada frente a antígenos del sistema nervioso central (SNC), liberados tras un daño por agentes neurotropos, o por analogía molecular3,4. Las manifestaciones neurológicas de EAD son frecuentemente polisintomáticas1,9, cerebrales y, con menor frecuencia, medulares. En niños4, son altamente sugestivas de EAD la alteración del nivel de consciencia o de comportamiento10, fiebre, meningismo o las crisis epilépticas1–3,11,12. En adultos es infrecuente y los síntomas más prevalentes son los motores, ataxia, sensitivos, de lenguaje y alteración del nivel de conciencia2,4,9,13. EAD suele ser monofásica3, aunque un 5,5-25% cursan con recaídas10. Estas pueden ocurrir en la misma topografía inicial (EAD-RR), y para algunos autores, en otras localizaciones (EAD multifásica)1,2,4,10. Ambas, en especial la forma de EAD multifásica, son objeto de amplia controversia, dado que el diagnóstico diferencial con EM3,10,11 conlleva implicaciones pronósticas y terapéuticas4.
Radiológicamente, en la EAD suelen observarse lesiones más extensas en la sustancia blanca cerebral que las de EM1,9. La captación de gadolinio es variable3, pero una captación simultánea apunta más hacia EAD9. Las lesiones de la sustancia gris, fundamentalmente en los ganglios basales, pueden ser bilaterales y aparecen hasta en el 60% de los casos9, mientras que en el primer episodio desmielinizante de EM son más infrecuentes. La presencia de «dedos de Dawson» en secuencias potenciadas en T1 debe orientar hacia un diagnóstico alternativo a la EAD3,11. El líquido cefalorraquídeo (LCR) puede ser normal o con pleocitosis linfocitaria e hiperproteinorraquia1. Las bandas oligoclonales (BOC) suelen estar ausentes o desaparecer durante el seguimiento, al contrario de lo descrito en EM1. Histopatológicamente, la EAD difiere de la EM, observándose «manguitos» de desmielinización perivenular con un prominente infiltrado inflamatorio, a expensas de macrófagos. Los márgenes de las lesiones están poco definidos14,15. Habitualmente, los pacientes son tratados con metilprednisolona intravenosa (MPIV) a altas dosis3 durante el episodio agudo, y en casos severos, sin respuesta a corticoides, la plasmaféresis16,17 o las inmunoglobulinas por vía intravenosa18 son alternativas terapéuticas9. El pronóstico de la EAD monofásica es variable; el 37-81% de los pacientes presentan resolución total de los síntomas2,11. La mortalidad (5-25%) se asocia a fallo respiratorio por afectación bulbar4. Hay poca experiencia respecto al tratamiento de EAD-RR, algunos autores han propuesto inmunosupresores12, ciclofosfamida2,11,12 o mitoxantrona4,12.
Por otra parte, la neuromielitis óptica (NMO) o enfermedad de Devic se caracteriza clásicamente por episodios de neuritis óptica y mielitis recurrentes, generalmente discapacitantes15,19. Infrecuentemente, la NMO puede cursar con síntomas bulbares, como hiperémesis y disfunción respiratoria, o incluso presentarse como una mielitis transversa20. En la RM de la NMO, las lesiones medulares suelen ser extensas, de más de 3 segmentos vertebrales18,20 y, en la mayoría de los casos, la sustancia blanca cerebral es normal. La determinación del anticuerpo anti-aquaporina 4 en 200421 ha permitido la separación de esta entidad de la EM y detectar formas intermedias o «espectro de NMO», cuya evolución está aún por determinar19,20. Sin embargo, los falsos negativos pueden llegar al 30-45%, especialmente en los casos tratados con fármacos inmunosupresores19. Durante el episodio agudo, la MTPIV20 y ocasionalmente plasmaféresis17 o inmunoglobulinas intravenosas (IVIG)18 son opciones terapéuticas. Como en la EAD-RR, no existen ensayos clínicos aleatorizados sobre el tratamiento de NMO recurrente, para la prevención de recaídas1,20.
ObjetivoEl objetivo es describir las características clínico-radiológicas de una paciente de 21 años con rombencefalomielitis recidivante seguida durante 9 años y discutir las alternativas diagnósticas y las consideraciones terapéuticas.
Paciente y métodosMujer de 21 años, sin antecedentes de enfermedad sistémica o autoinmunitaria, diagnosticada de gastritis en el servicio de urgencias en febrero de 2003, por un cuadro de 2 días de evolución de vómitos, febrícula y dolor mesogástrico. Ocho días más tarde asoció dificultad progresiva para deglutir, odinofagia y vómitos incoercibles. Una TC toracoabdominal y una endoscopia urgentes, fueron normales.
Posteriormente, la paciente volvió a ser valorada por disnea, sensación de cuerpo extraño faríngeo, dificultad fonatoria y atragantamiento, junto con dolores lancinantes y disestesia dolorosa en banda ancha en la zona dorsolumbar. No refería antecedentes de vacunaciones o infecciones previas a este cuadro. La exploración física general fue normal. Al ser explorada por neurología, se observó limitación para la abducción del ojo izquierdo y nistagmo multidireccional en todas las posiciones de la mirada, abolición del reflejo nauseoso, importante disartria y dificultad respiratoria. La maniobra de Barré era positiva en el miembro superior derecho y los reflejos osteotendinosos estaban hipoactivos. Tenía una banda de hipoalgesia a nivel D4-L1, sensibilidad proprioceptiva alterada en los cuatro miembros y dismetría en el miembro superior izquierdo. La agudeza visual y el fondo de ojo eran normales.
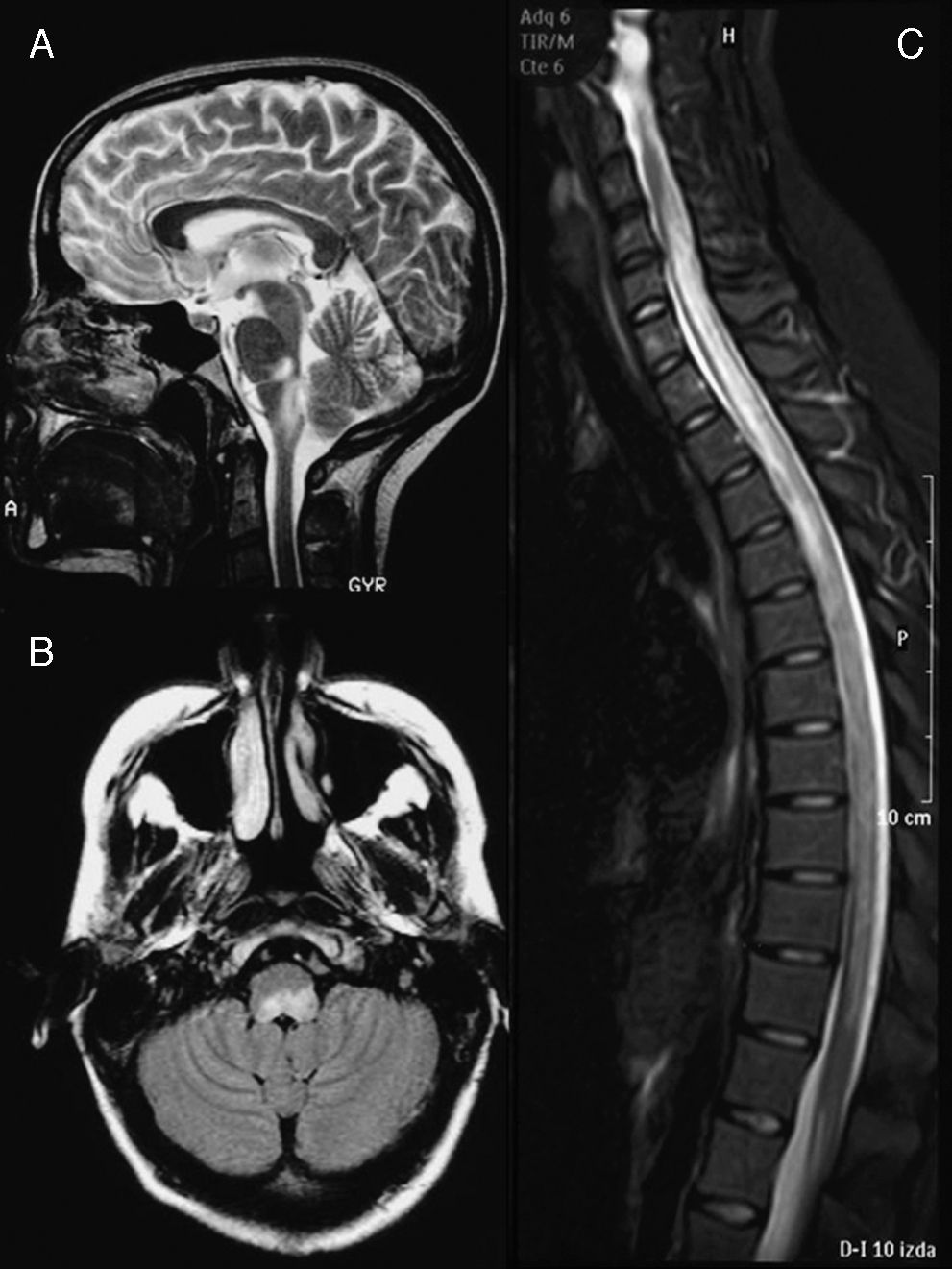
La RM craneal (fig. 1A) mostró lesiones hiperintensas en secuencias T2 en los dos tercios del bulbo dorsal con compresión del cuarto ventrículo y pedúnculo cerebeloso izquierdo. En la RM medular (fig. 1B), se apreciaba una banda hiperintensa en secuencias potenciadas en T2, desde C4 a L1. El LCR mostró pleocitosis (50/μl) de predominio monocítico, estéril, proteínas 62mg/dl y glucosa 84mg/dl. El índice IgG fue normal y no se realizaron BOC. El estudio microbiológico descartó las etiologías infecciosas (Borrelia, Brucella, Mycoplasma, parvovirus, citomegalovirus, virus herpes, virus de la varicela-zóster, virus de Epstein-Barr, toxoplasmosis, enterovirus, micobacterias, virus de la inmunodeficiencia adquirida, virus de la hepatitis C y VHB, Listeria, gram, auramina). El estudio serológico descartó causas autoinmunitarias o tumorales.
. Corte sagital en secuencias T2 (A) y axial FLAIR (B) en los que se observa una alteración de la señal a nivel del bulbo dorsal, que ejerce efecto masa sobre el IV ventrículo. C) Imagen de resonancia medular, en secuencia STIR sin contraste, en la que se observa una extensa alteración de señal desde C4 hasta L1, con ligero efecto masa.")
Imagen de RM cerebral en el momento del diagnóstico (febrero de 2003). Corte sagital en secuencias T2 (A) y axial FLAIR (B) en los que se observa una alteración de la señal a nivel del bulbo dorsal, que ejerce efecto masa sobre el IV ventrículo. C) Imagen de resonancia medular, en secuencia STIR sin contraste, en la que se observa una extensa alteración de señal desde C4 hasta L1, con ligero efecto masa.
El diagnóstico inicial fue rombencefalomielitis autoinmunitaria. La paciente ingresó en unidad de cuidados intensivos por distrés respiratorio secundario a neumonía aspirativa. Fue tratada con antibioterapia de amplio espectro y MPIV durante 5 días. Debido a la respuesta escasa a corticoides, se trató con 5 ciclos de plasmaféresis seguido de IVIG a dosis de 0,4mg/kg/día durante 5 días. Al alta, un mes tras el ingreso, presentaba una paresia del VI par craneal izquierdo, nistagmus multidireccional y una marcha inestable, autónoma.
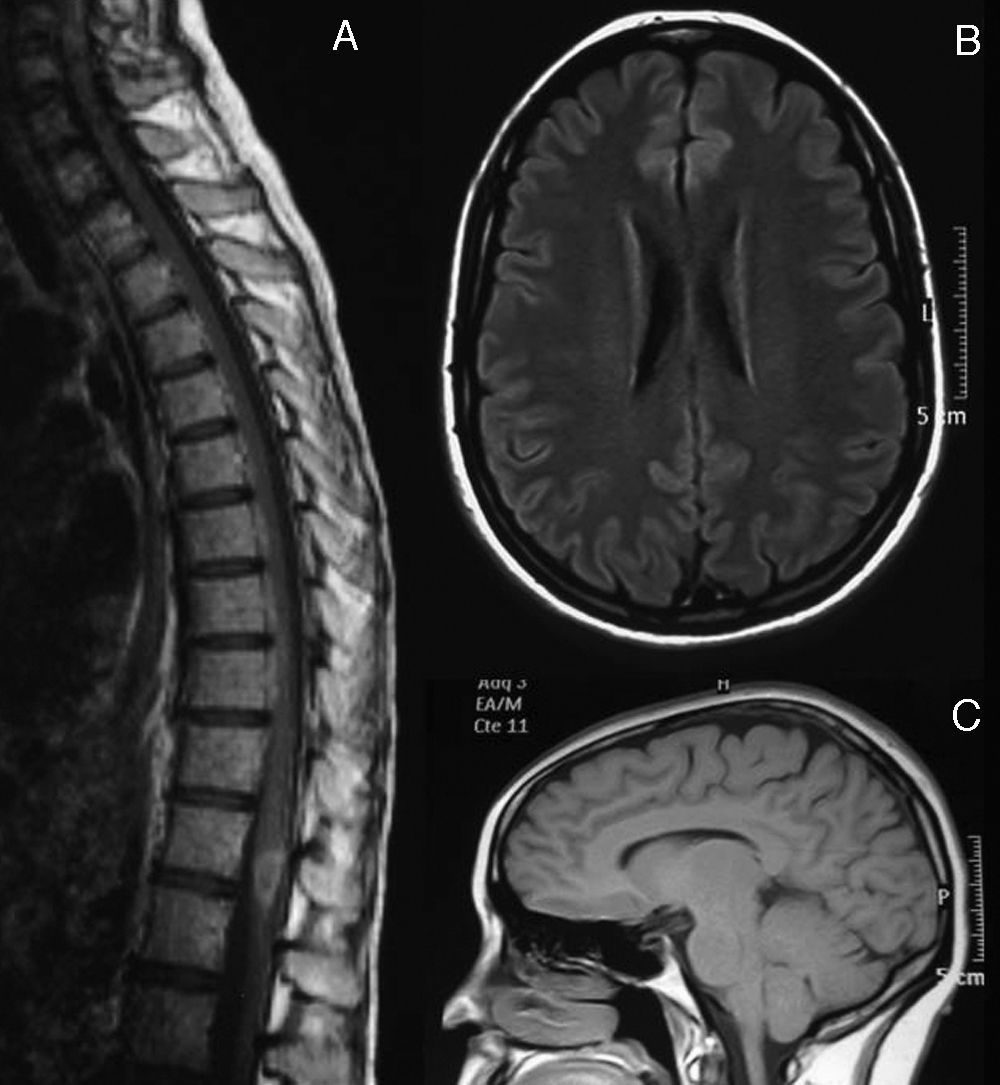
Con posterioridad, la paciente volvió a presentar 7 recaídas espontáneas (tabla 1). De las 7, 4 han consistido en mielitis de gravedad leve-moderada con afectación motora y/o sensitiva, todas con nivel dorsal, con una recuperación tras unos días. La primera recaída coincidió con retirada de los glucocorticoides. Se instauró tratamiento con azatioprina. Las restantes consistieron en dolor paroxístico bilateral en hemicinturón asociados a hipoalgesia metamérica D5-D10 bilateral. Durante el curso clínico los potenciales evocados visuales fueron normales y la serología para HTLV-1 y anti-aquaporina 4, realizadas durante una de las recaídas (mayo 2007), fueron negativas. En la RM, la lesión medular presentó captación de gadolinio durante varias de las recaídas clínicas y no se observaron otras lesiones medulares o craneales, o signos de daño axonal (fig. 2).
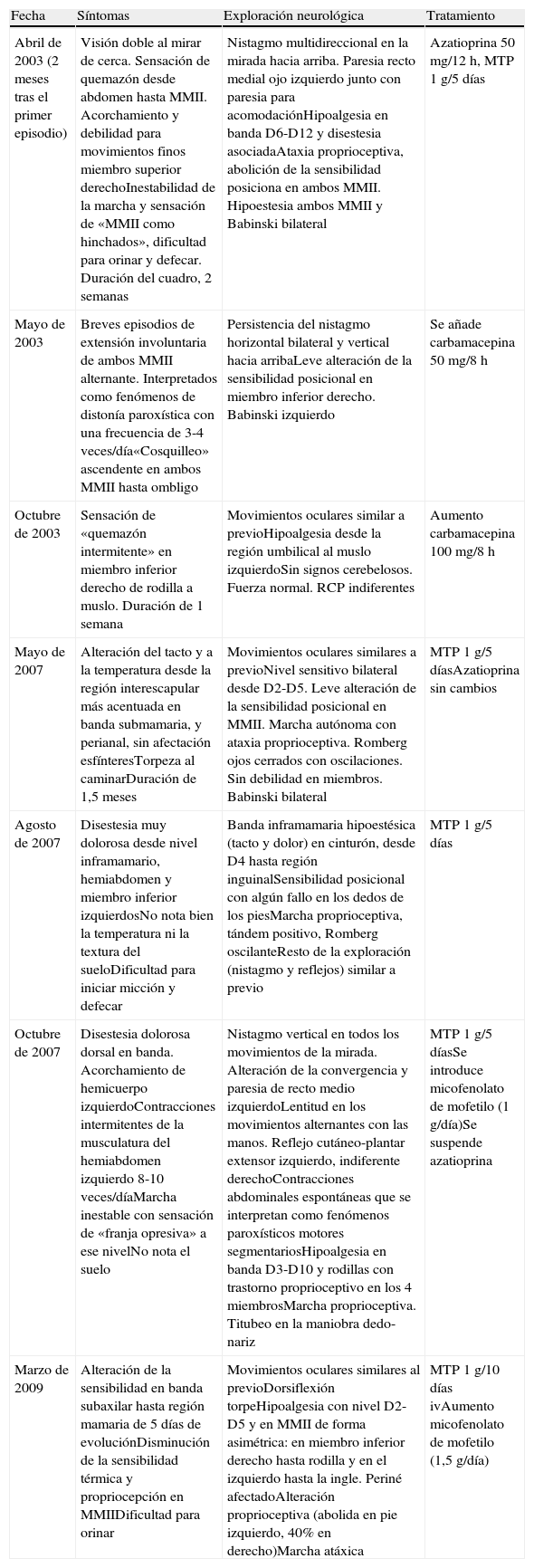
Recidivas clínicas y tratamiento
| Fecha | Síntomas | Exploración neurológica | Tratamiento |
| Abril de 2003 (2 meses tras el primer episodio) | Visión doble al mirar de cerca. Sensación de quemazón desde abdomen hasta MMII. Acorchamiento y debilidad para movimientos finos miembro superior derechoInestabilidad de la marcha y sensación de «MMII como hinchados», dificultad para orinar y defecar. Duración del cuadro, 2 semanas | Nistagmo multidireccional en la mirada hacia arriba. Paresia recto medial ojo izquierdo junto con paresia para acomodaciónHipoalgesia en banda D6-D12 y disestesia asociadaAtaxia proprioceptiva, abolición de la sensibilidad posiciona en ambos MMII. Hipoestesia ambos MMII y Babinski bilateral | Azatioprina 50 mg/12 h, MTP 1 g/5 días |
| Mayo de 2003 | Breves episodios de extensión involuntaria de ambos MMII alternante. Interpretados como fenómenos de distonía paroxística con una frecuencia de 3-4 veces/día«Cosquilleo» ascendente en ambos MMII hasta ombligo | Persistencia del nistagmo horizontal bilateral y vertical hacia arribaLeve alteración de la sensibilidad posicional en miembro inferior derecho. Babinski izquierdo | Se añade carbamacepina 50 mg/8 h |
| Octubre de 2003 | Sensación de «quemazón intermitente» en miembro inferior derecho de rodilla a muslo. Duración de 1 semana | Movimientos oculares similar a previoHipoalgesia desde la región umbilical al muslo izquierdoSin signos cerebelosos. Fuerza normal. RCP indiferentes | Aumento carbamacepina 100 mg/8 h |
| Mayo de 2007 | Alteración del tacto y a la temperatura desde la región interescapular más acentuada en banda submamaria, y perianal, sin afectación esfínteresTorpeza al caminarDuración de 1,5 meses | Movimientos oculares similares a previoNivel sensitivo bilateral desde D2-D5. Leve alteración de la sensibilidad posicional en MMII. Marcha autónoma con ataxia proprioceptiva. Romberg ojos cerrados con oscilaciones. Sin debilidad en miembros. Babinski bilateral | MTP 1 g/5 díasAzatioprina sin cambios |
| Agosto de 2007 | Disestesia muy dolorosa desde nivel inframamario, hemiabdomen y miembro inferior izquierdosNo nota bien la temperatura ni la textura del sueloDificultad para iniciar micción y defecar | Banda inframamaria hipoestésica (tacto y dolor) en cinturón, desde D4 hasta región inguinalSensibilidad posicional con algún fallo en los dedos de los piesMarcha proprioceptiva, tándem positivo, Romberg oscilanteResto de la exploración (nistagmo y reflejos) similar a previo | MTP 1 g/5 días |
| Octubre de 2007 | Disestesia dolorosa dorsal en banda. Acorchamiento de hemicuerpo izquierdoContracciones intermitentes de la musculatura del hemiabdomen izquierdo 8-10 veces/díaMarcha inestable con sensación de «franja opresiva» a ese nivelNo nota el suelo | Nistagmo vertical en todos los movimientos de la mirada. Alteración de la convergencia y paresia de recto medio izquierdoLentitud en los movimientos alternantes con las manos. Reflejo cutáneo-plantar extensor izquierdo, indiferente derechoContracciones abdominales espontáneas que se interpretan como fenómenos paroxísticos motores segmentariosHipoalgesia en banda D3-D10 y rodillas con trastorno proprioceptivo en los 4 miembrosMarcha proprioceptiva. Titubeo en la maniobra dedo-nariz | MTP 1 g/5 díasSe introduce micofenolato de mofetilo (1 g/día)Se suspende azatioprina |
| Marzo de 2009 | Alteración de la sensibilidad en banda subaxilar hasta región mamaria de 5 días de evoluciónDisminución de la sensibilidad térmica y propriocepción en MMIIDificultad para orinar | Movimientos oculares similares al previoDorsiflexión torpeHipoalgesia con nivel D2-D5 y en MMII de forma asimétrica: en miembro inferior derecho hasta rodilla y en el izquierdo hasta la ingle. Periné afectadoAlteración proprioceptiva (abolida en pie izquierdo, 40% en derecho)Marcha atáxica | MTP 1 g/10 días ivAumento micofenolato de mofetilo (1,5 g/día) |
MMII: miembros inferiores; MMSS: miembros superiores; MTP: metilprednisolona; ROT: reflejos osteotendinosos.
 RM medular con gadolinio intravenoso. Se observa una lesión hipodensa en secuencias T1 desde C2 hasta D10, con captación de contraste a nivel D8-D9. A nivel cerebral, en secuencias FLAIR axial (B) y T1 sagital (C), no se observan lesiones de sustancia blanca ni signos de atrofia.")
Agosto de 2007. A) RM medular con gadolinio intravenoso. Se observa una lesión hipodensa en secuencias T1 desde C2 hasta D10, con captación de contraste a nivel D8-D9. A nivel cerebral, en secuencias FLAIR axial (B) y T1 sagital (C), no se observan lesiones de sustancia blanca ni signos de atrofia.
La paciente fue tratada de forma crónica con azatioprina a dosis de 50mg/12h y, dada la persistencia de recaídas, se sustituyó la azatioprina por micofenolato de mofetilo a dosis de 1-1,5g/día. En la actualidad, la paciente lleva 3 años sin recaídas, camina y salta normalmente, y a la exploración sólo persiste un nistagmo vertical hacia arriba y horizontal bilateral sin interferencia funcional.
DiscusiónNuestro caso sugiere una forma de rombencefalomielitis recurrente compatible con EAD-RR por criterios clínicos y radiológicos, con buena evolución clínica y respuesta terapéutica a inmunosupresores. Actualmente, la paciente está clínicamente asintomática tras 7 recaídas, a pesar de haber presentado en 3 de ellas incapacidad moderada. Es destacable que todas las recaídas tuvieron, aunque con distinta severidad, la misma localización que el evento inicial y sólo la primera recaída puede atribuirse a la retirada paulatina de los glucocorticoides.
Las imágenes en la RM cerebral y medular son extensas, con engrosamiento medular, similar a lo descrito en casos excepcionales de EAD2,10, e inusuales para EM. Al menos en dos ocasiones, coincidiendo con una exacerbación de los síntomas, las imágenes medulares captaban contraste. Es llamativo que tras 7 recidivas de la enfermedad no se observen datos de necrosis medular ni agujeros negros en la RM cerebral, lo que sugiere ausencia de lesión axonal, más propio de la EM. La evolución del caso y los datos expuestos excluyen el diagnóstico de EM22.
El límite con la NMO es todavía más controvertido23. Desde el punto de vista clínico, nuestra paciente en ningún momento de la evolución ha tenido sintomatología visual propia de la NMO y los potenciales evocados han sido normales. En la NMO es rara la recuperación completa de los episodios sin discapacidad15,19,23,24. Aunque las imágenes medulares de nuestra paciente asemejan a las descritas en NMO24, no muestran datos de necrosis tisular, como es habitual en la NMO15,24, aunque recientemente se han descrito casos en los que las alteraciones radiológicas pueden mejorar entre los episodios o incluso desaparecer20. La determinación de IgG frente a AQP4 fue negativa, aunque la determinación se realizó bajo tratamiento inmunosupresor, por lo que no podemos confirmar de forma definitiva que nuestra paciente corresponda a una forma no común de NMO recurrente según los criterios vigentes20,24.
La evolución clínica y la negatividad de las pruebas microbiológicas e inmunológicas realizadas al inicio y durante varios de los episodios, excluyen otras entidades que pueden mimetizar estos síntomas.
Respecto al tratamiento, no existe un consenso claro. Durante el brote inicial, la paciente fue tratada con MP IV a dosis altas, y debido a la severidad del brote y a la respuesta incompleta se trató con plasmaféresis e IVIG, con mejoría clínica marcada16,17. Se instauró tratamiento con azatioprina a raíz de la primera recidiva, al estar descrito casos de buena respuesta a este fármaco12,24,25. Los brotes persistieron, por lo que se optó por cambiar a micofenolato de mofetilo, útil en otras enfermedades autoinmunitarias, incluidas la NMO y la EM25 al inducir la apoptosis de células T reactivas y disminuir la reactividad humoral26. Desde hace 2 años y medio está asintomática.
ConclusionesNuestro estudio ilustra que en determinados casos de enfermedades desmielinizantes, la caracterización diagnóstica no es fácil pese a los criterios vigentes. Consideramos que nuestro caso comparte datos clínicos y radiológicos de EAD-RR de comienzo en el adulto y de una forma inusual de NMO. El excelente pronóstico a medio plazo con tratamiento inmunosupresor sugiere más una forma poco común de EAD-RR. El tratamiento con micofenolato de mofetilo podría ser una terapia alternativa para estos pacientes.
Conflicto de interesesLos autores declaran no tener conflictos de intereses.

