




La enfermedad de Erdheim-Chester (EEC) es una histiocitosis infrecuente, de células no Langerhans, que se presenta fundamentalmente en la edad media de la vida y que puede afectar a ambos sexos.
Caso clínicoPresentamos el caso de un varón de 52 años con EEC sistémica y lesión tentorial casual en estudio radiológico. Se realizó biopsia de la lesión, que confirmó el diagnóstico. Debido a la afectación sistémica, recibió tratamiento con interferón α y anakinra, sin respuesta. En controles sucesivos, el paciente no presenta clínica neurológica y la lesión se mantiene estable en las pruebas radiológicas. Se muestran las imágenes de la resonancia magnética, se discuten las opciones diagnósticas y se muestra un campo representativo del estudio de anatomía patológica.
DiscusiónLa EEC es una enfermedad rara, que afecta principalmente al sistema osteoarticular, aunque también a los sistemas cardiovascular y respiratorio, y la región retroperitoneal, y que compromete el sistema nervioso central con frecuencia. Nuestro caso tiene la peculiaridad de la afectación meníngea. Se realiza una revisión bibliográfica de los casos descritos hasta la actualidad y se discuten las mejores opciones terapéuticas, analizando los diferentes diagnósticos diferenciales posibles.
Erdheim-Chester disease (ECD) is a rare, non-Langerhans histiocytosis. It is most often diagnosed in the middle age, with an average age at onset of 53 years. It can affect men and women.
Case reportWe report the case of a 52-year old man with systemic ECD and an incidental tentorial lesion found in a radiological study. A brain biopsy confirmed the diagnosis, and he was treated conservatively with successive controls, given the absence of neurological symptoms. We present MRI images and pathology slides and discuss the disease diagnosis.
DiscussionECD is a rare disease, which affects mainly the osteoarticular system but also the cardiovascular and respiratory systems and the retroperitoneal space, and that often compromises the Central Nervous System. Our patient presents meningeal involvement. We conduct a literature review of the cases described until the present day, discuss the best therapeutic options and analyze, the potential differential diagnoses.
La enfermedad de Erdheim-Chester (EEC) fue descrita por primera vez en 1930, como una forma rara de histiocitosis de células no Langerhans, de origen desconocido y afectación multisistémica. Se caracteriza por infiltración de histiocitos espumosos cargados de lípidos en diversos tejidos. La presencia de marcadores inmunohistoquímicos (CD68+/CD1a–) y la ausencia de proteína S y gránulos de Birbeck permiten su diferenciación del resto de las histiocitosis1,2.
Hasta hoy se han publicado menos de 600 casos, diagnosticados en personas de todas las edades, siendo más frecuente en adultos, con una media de edad de 53 años. La manifestación clínica más frecuente es el dolor, por la afectación ósea, en forma de lesiones esclerosantes en diáfisis y metáfisis de huesos largos de extremidades inferiores, aunque no son infrecuentes las alteraciones endocrinológicas, oftalmológicas, cardiovasculares, pulmonares y del sistema nervioso central (SNC)3,4. La afectación meníngea es nodular, simulando el aspecto de los meningiomas5.
Caso clínicoPresentamos el caso de un varón de 52 años, sin antecedentes familiares ni personales, que comenzó en 2011 con la aparición de xantelasmas palpebrales bilaterales, que fueron tratados quirúrgicamente. Dos años después, desarrolló gonalgia bilateral, observando alteraciones osteoescleróticas en ambas rodillas en los distintos estudios radiológicos realizados. Ante la persistencia del dolor, el paciente fue sometido a una gammagrafía ósea con Tc99m, cuyo resultado demostró un aumento moderado y simétrico de la captación del radiotrazador en ambas rodillas, así como en las epífisis distales de ambas tibias.
Ante dichos resultados, el paciente fue sometido a una biopsia ósea, en la que destacó el aumento de histiocitos CD68+, CD1a– y S-100–, con una marcada fibrosis y un patrón claro de osteosclerosis. Tras estos hallazgos histopatológicos, y ante la sospecha de EEC, se decidió realizar ampliación del estudio con una tomografía computarizada de tórax, abdomen y pelvis, en la cual se pudo observar un aumento de la densidad perirrenal compatible con fibrosis, lo que originaba una hidronefrosis grado ii. Se observó también que dicha fibrosis se extendía hacia ambos espacios pararrenales posteriores, con mesenteritis.
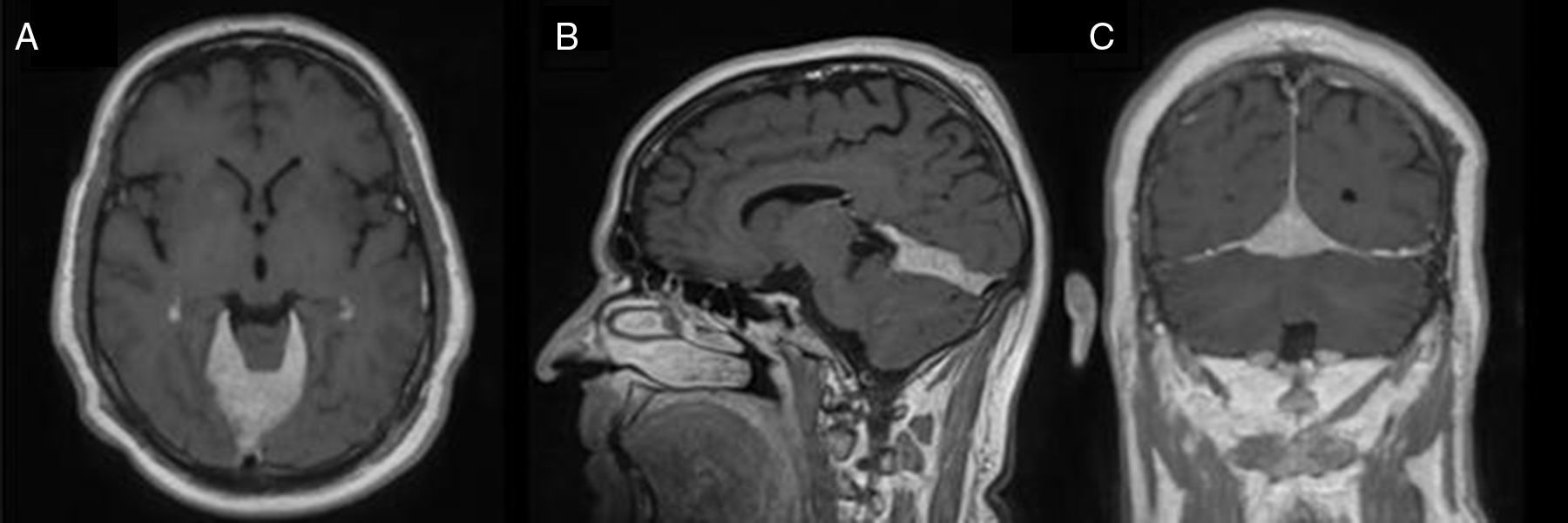
Ante los hallazgos radiológicos sistémicos y la presencia de alteraciones hormonales en el estudio bioquímico, se realizó una resonancia magnética (RM) cerebral para descartar una posible afectación del SNC. En la RM cerebral (fig. 1) se visualizó una gran masa extraaxial, a nivel tentorial, descrita como un engrosamiento difuso y polilobulado del tentorio, fundamentalmente en línea media y región derecha de la misma, con unos diámetros máximos de 2cm en el eje cráneo-caudal, 3,3cm en el diámetro transverso y 5cm en el eje antero-posterior, con captación homogénea de contraste.
, sagital (B) y coronal (C), que muestran lesión extraaxial en el tentorio, de predominio en línea media, que realza de forma homogénea tras la administración de contraste paramagnético.")
Ante la imposibilidad de llevar a cabo una resección completa por su localización, y dado el hallazgo casual de la imagen, se decidió realizar una craneotomía suboccipital y una biopsia de la lesión, con el objetivo de confirmar una posible afectación meníngea por EEC y descartar otras posibles opciones diagnósticas, como la de un meningioma en placa.
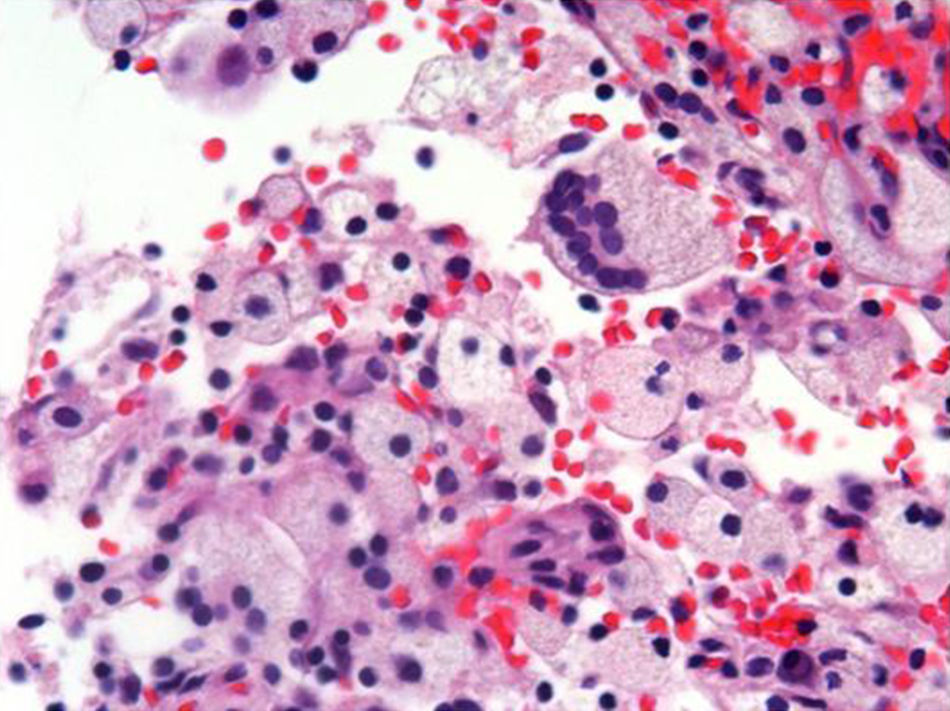
El resultado del estudio histopatológico reveló la presencia de acúmulos nodulares de histiocitos de carácter espumoso, uni o multinucleados (fig. 2), cuyo estudio inmunohistoquímico resultó compatible con EEC. El estudio genético fue positivo para la mutación V600E del gen BRAF.
.")
Debido a la afectación sistémica que presentó el paciente, recibió tratamiento con interferón α y posteriormente con anakinra, sin respuesta a ambos tratamientos. Desde el punto de vista, neurológico se ha mantenido asintomático y con estabilidad radiológica en los sucesivos controles realizados.
DiscusiónLa EEC es una rara entidad, de origen desconocido y afectación sistémica por histiocitos no Langerhans, que afecta a población de ambos sexos, con discreto predominio en varones. La edad media de presentación es entre la cuarta y la séptima décadas de la vida, aunque también se observan casos en edades pediátricas1-3.
Las manifestaciones clínicas son muy variadas y dependen de las localizaciones comprometidas por la enfermedad4-6. La mayoría de los pacientes con EEC presentan afectación ósea en el momento del diagnóstico y la gran mayoría de los mismos tendrá al menos una localización de afectación no ósea. La clínica de presentación más frecuente es el dolor óseo, de predominio habitualmente en miembros inferiores, los síntomas neurológicos, la diabetes insípida y los síntomas constitucionales7,8.
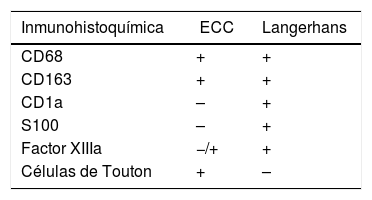
El diagnóstico de la EEC puede resultar difícil debido a que es una enfermedad poco común y sus manifestaciones clínicas son muy variadas6. Los criterios diagnósticos empleados están basados en los hallazgos clínicos, radiológicos e histopatológicos, siendo este último el factor más importante. Desde el punto de vista del estudio anatomopatológico, se debe realizar una biopsia de las lesiones osteoescleróticas, cuando esto sea posible. El estudio del tejido afectado muestra infiltración por histiocitos de carácter espumoso no Langerhans, sin gránulos de Birbeck, originando en su entorno una reacción fibrosante. En algunas ocasiones se observan células gigantes de Touton asociadas. Los estudios inmunohistoquímicos nos permiten diferenciar la EEC de la enfermedad de Langerhans8,9 (tabla 1). También se han descrito casos de EEC asociado a histiocitosis de células de Langerhans (histiocitosis mixtas), que podrían estar en relación con la mutación BRAFV600E9.
Junto a los datos histopatológicos debemos considerar la presencia de los hallazgos radiológicos más frecuentemente asociados, como es la osteosclerosis cortical bilateral en diáfisis y metáfisis de huesos de miembros inferiores10.
Las manifestaciones clínicas de la EEC varían según el órgano afectado, con lesiones osteoarticulares, valvulopatías cardíacas, pericarditis, afectación de los grandes vasos, neumopatías intersticiales y afectación retroperitoneal1-5,7,8.
La afectación de la EEC en el SNC varía entre el 25 y el 50% de los pacientes. Las manifestaciones clínicas pueden deberse a la presencia de lesiones a lo largo de todo el neuroeje y de estructuras adyacentes tales como meninges, órbita o huesos de la cara11. Los síntomas más frecuentes son diabetes insípida, exoftalmos, ataxia cerebelosa, panhipopituitarismo y edema de papila12,13. Otras manifestaciones clínicas más raras son la afectación de los huesos de la cara, la infiltración de arterias cerebrales, la afectación del seno sagital o del plexo coroideo y la formación de tumores14,15. La afectación única en el SNC es rara; en el 66% de los pacientes se desarrolla infiltración de 2 o más localizaciones simultáneamente. La sintomatología neurológica conlleva el desarrollo de incapacidades importantes, siendo un factor determinante en la evolución fatal de estos pacientes16,17.
La afectación meníngea de esta enfermedad es nodular, simulando la aparición de meningiomas en placa. Estas lesiones en la RM cerebral se comportan como hipointensas en secuencias potenciadas en T2 e isointensas en T1, con captación intensa de gadolinio11.
La EEC es una enfermedad rara, con afectación de múltiples sistemas, que nos obliga a un manejo multidisciplinar tanto en su diagnóstico como en su tratamiento. Actualmente, es necesario realizar en este tipo de pacientes un estudio amplio de extensión de la enfermedad, para comprender y manejar las diferentes afecciones en los diversos órganos. La afectación a nivel del SNC en estos pacientes es muy heterogénea, siendo necesario el desarrollo de nuevos protocolos terapéuticos para conseguir mejores resultados. La cirugía resulta útil tanto para filiar la enfermedad, como para extirpar las lesiones que originen sintomatología neurológica.
Desde el punto de vista del tratamiento médico, y dado el escaso número de casos publicados sobre esta enfermedad, no existen protocolos concluyentes. Actualmente, el tratamiento más empleado en estos pacientes es el interferón α, el cual consigue la estabilización de la enfermedad en muchos casos18. En una serie de 53 pacientes tratados con interferón, en un estudio prospectivo, no aleatorizado y con corte observacional, se observó mejor respuesta que con el resto de las terapias en términos de supervivencia, siendo esta una variable independiente19. Las mejores respuestas al tratamiento se observan a nivel de piel, SNC, pulmón y corazón.
Como alternativas a este tratamiento, se ha empleado anakinra, un antagonista del receptor IL-1, con buenos resultados en un pequeño número de pacientes20. Este fármaco es muy útil para el control del dolor óseo y de los síntomas constitucionales, siendo ineficaz para limitar la afectación neurológica. Actualmente, se están empleando otros fármacos para el control de la enfermedad, tales como infliximab, tocilizumab, cladribina o vemurafenib21-23.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
FinanciaciónNo hemos recibido financiación para su elaboración
Conflicto de interesesNo existen conflictos de intereses.
