





Describir los hallazgos clínicos, electrofisiológicos y moleculares en un caso de parálisis periódica hipocalémica tipo 2 (PPH2) y una nueva mutación no descripta en el gen SCN4A.
Caso clínicoPaciente de 32 años, en seguimiento por episodios de debilidad muscular súbita de inicio vespertino desde los 16 años. Recibió acetazolamida con agravamiento de sus síntomas. Patrón electrofisiológico tipo iv. Se decide secuenciación de SCN4A (variante en heterocigosis en exón 5, c.664C>G; p.Arg222Gly), no descripta previamente y predictiva de ser patogénica.
ConclusionesLa caracterización electrofisiológica y molecular aporta información relevante en las PPH2, ya que puede predecir la respuesta farmacológica.
To describe clinical, electrophysiological and molecular findings in a case of type 2 hypokalemic periodic paralysis (PPH2) and a novel mutation in SCN4A gene, which had not described to date.
Case report32 year-old male patient in follow-up for episodes of sudden muscle weakness, beginning at 16 years of age. Patient received acetazolamide with worsening symptoms. Electrophysiological pattern type iv. It was decided to perform SCN4A sequencing (heterozygous variant in exon 5, c.664C>G); p.Arg222Gly, which had not been previously described and which is predictive of pathogenesis.
ConclusionsElectrophysiological and molecular characterization provides relevant information on PPH2, as it can predict patient's response to drug treatment.
Las canalopatías musculares son un conjunto de enfermedades de etiología genética, condicionadas por mutaciones en canales iónicos dependientes del voltaje. Abarcan diferentes categorías, como ciertos síndromes miotónicos, las parálisis periódicas y la hipertermia maligna, entre otras. Las manifestaciones clínicas cardinales incluyen la dificultad para la relajación muscular (miotonía) o la presencia de debilidad muscular transitoria o permanente. Las parálisis periódicas constituyen un subgrupo de estas enfermedades que poseen como rasgo distintivo episodios recurrentes y reversibles de debilidad muscular prolongada asociados a la presencia o no de miotonía y progresión a debilidad muscular permanente. Las mismas pueden ser clasificadas en hipercalémicas e hipocalémicas, según los niveles séricos de potasio durante las crisis. Las parálisis hipocalémicas son de herencia autosómica dominante y responden a mutaciones principalmente en 3 genes: CACNA1S (variante tipo i: 60% de los casos) y SCN4A (variante tipo ii: 20% de los casos) y en menor medida KCNJ18 (3,5%). Su diagnóstico se apoya en criterios clínicos-serológicos, electrofisiológicos y moleculares1,2.
Las características clínicas de las parálisis hipocalémicas incluyen episodios paroxísticos transitorios y recurrentes de debilidad muscular flácida, en tronco y extremidades. Habitualmente cursa sin compromiso de la musculatura bulbar, ocular ni respiratoria, durante los cuales, mayormente, se encuentran dosajes de potasio bajos en sangre. Los episodios duran entre minutos y horas, llegando a alcanzar días, en ocasiones. No presentan fenómenos miotónicos interictales, rasgo que las diferencia de las parálisis periódicas hipercalémicas (estas últimas son también de carácter autosómico dominante con alta penetrancia, presentan preferentemente afección de cara, ojos y faringe, su duración es más breve, siendo, por lo general, de minutos a horas y las mutaciones se producen sobre los canales de sodio). Con el correr de los años, los pacientes pueden desarrollar una miopatía proximal fija en un 25% de los casos. Se han descripto como desencadenantes de las crisis: dieta rica en carbohidratos y sodio, estrés, descanso luego de ejercicio extremo, bajas temperaturas y ciertos fármacos (corticoides, insulina, betaagonistas). La sospecha diagnóstica se realiza sobre la base de la clínica, el hallazgo de hipocalemia durante los eventos y los antecedentes familiares positivos (si bien pueden existir mutaciones de novo). En general, las alternativas terapéuticas farmacológicas propuestas para disminuir la frecuencia y la intensidad de las crisis, como los diuréticos ahorradores de potasio, suelen ser efectivas a excepción del subtipo ii, en donde pueden agravarse3-5.
Actualmente, los protocolos electrofisiológicos, como el de Fournier et al., que analizan el comportamiento de la amplitud del potencial de acción en el tiempo, posterior un ejercicio corto (10 s) o largo (5 min), permiten determinar patrones que correlacionan con el fenotipo clínico el gen alterado y permiten, como mencionamos, estimar la respuesta terapéutica a los fármacos convencionales6. El objetivo de este trabajo es la caracterización clínica, electrofisiológica y molecular en un paciente con parálisis periódica hipocalémica tipo ii, así como la descripción de una variante probablemente patogénica en SCN4A, no descripta previamente.
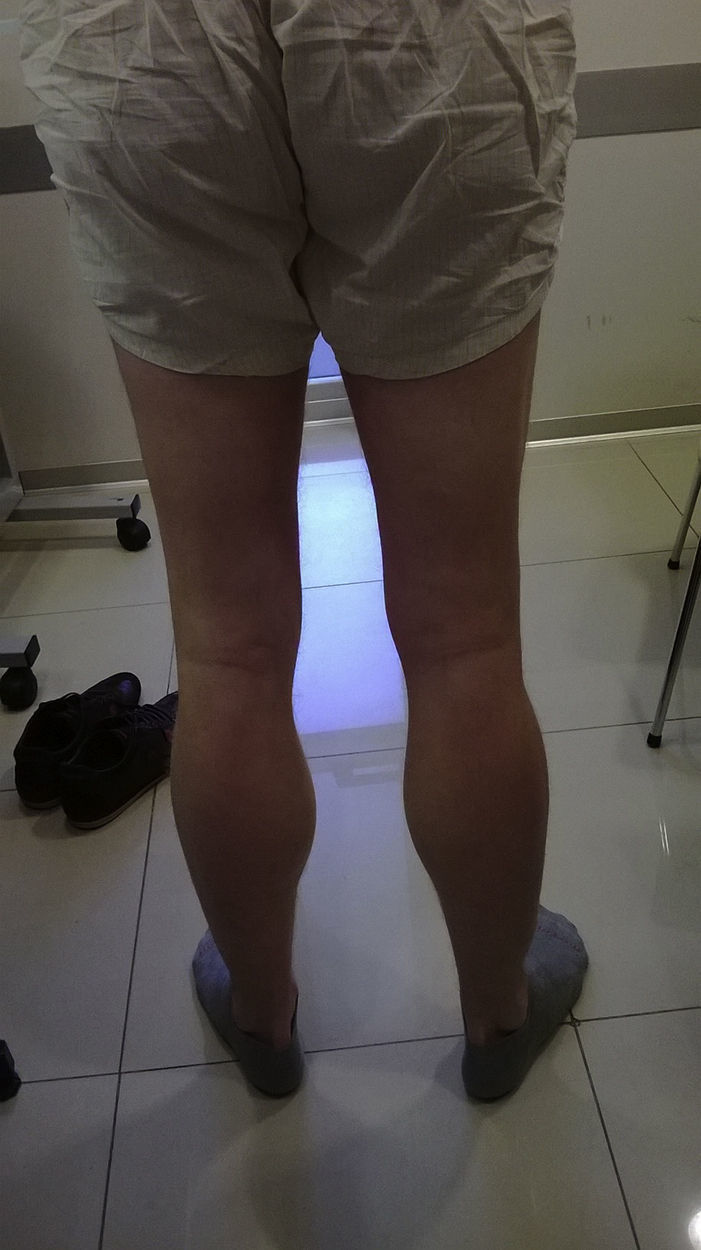
Caso clínicoSe describe el caso de un paciente masculino de 32 años de edad, hijo único, sin antecedentes familiares de relevancia, quien desde los 16 años presentaba episodios de pérdida de tono postural y debilidad muscular súbita, que iniciaba en las piernas, con progresión a extremidades y tronco, en ocasiones con dificultades para hablar y respirar, de inicio vespertino mayormente, que duraban de minutos a horas. Las crisis se habían agravado en últimos años, volviéndose semanales y de horas de duración. No había relación con actividad física, ayuno o consumo de alcohol. Con la evolución, comenzaron a ser diarias, algunas de ellas requiriendo traslado a centros médicos de urgencia en donde se le detectaron descensos marcados del potasio sérico (1,7mM/l en una oportunidad). Comenzó seguimiento neurológico en otro centro con diagnóstico probable de parálisis periódica hipocalémica, Se indicó dieta baja en carbohidratos y recibió acetazolamida como tratamiento farmacológico preventivo, la cual le produjo franco agravamiento de sus síntomas. Concurrió a nuestro centro para revaloración diagnóstica. En el examen neurológico: lúcido, sin trastornos del lenguaje, pares craneales conservados, fondo de ojo normal, sin debilidad muscular en las pruebas de fuerza segmentaria, hipertrofia gemelar desproporcionada al trofismo del resto de su musculatura (fig. 1). Tono muscular, sensibilidad, taxia, marcha y reflejos osteotendinosos, normales. En los estudios de laboratorio se observó una elevación discreta de la creatina-cinasa, con valores de ionograma, perfil tiroideo y función renal conservados.

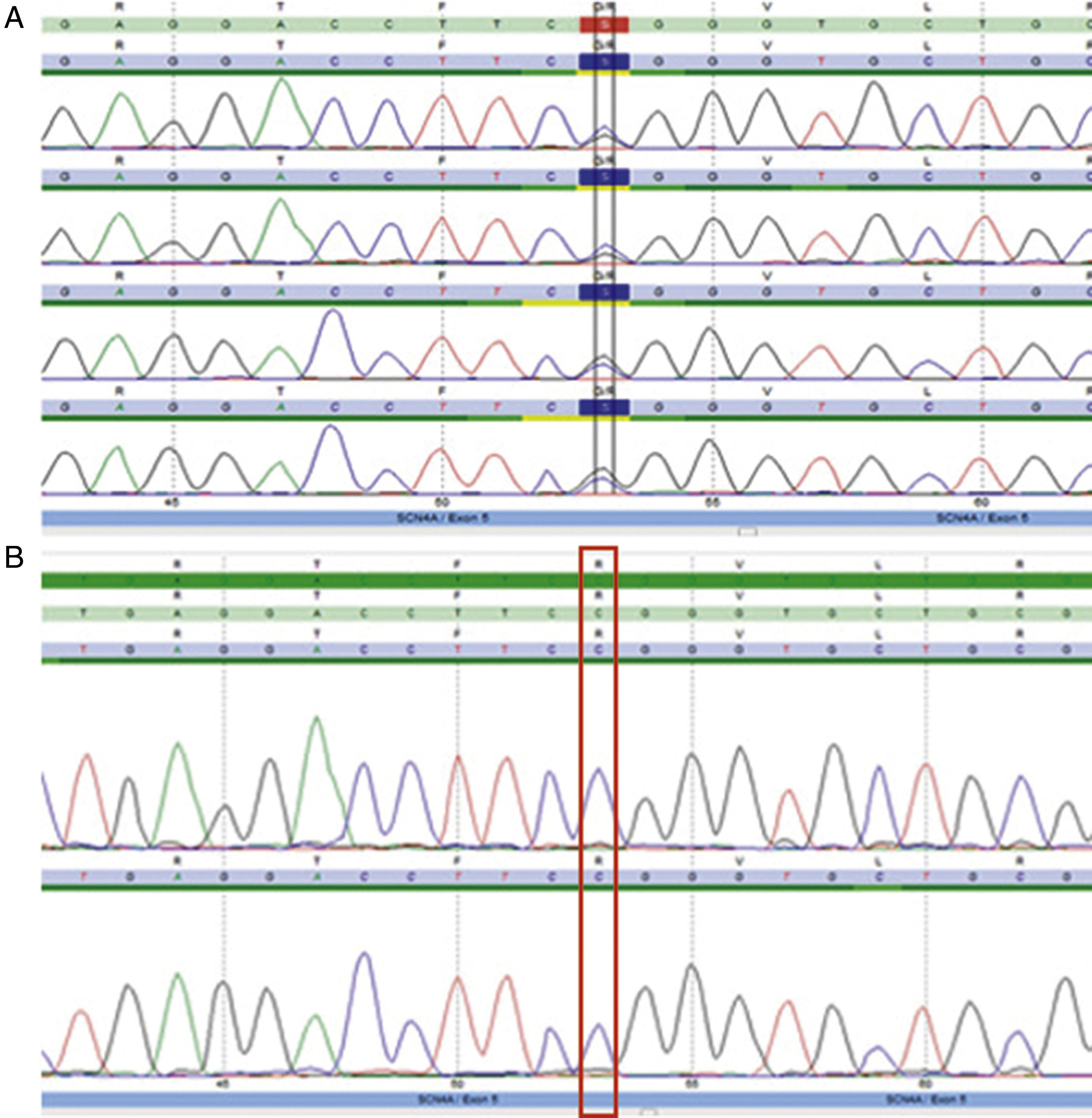
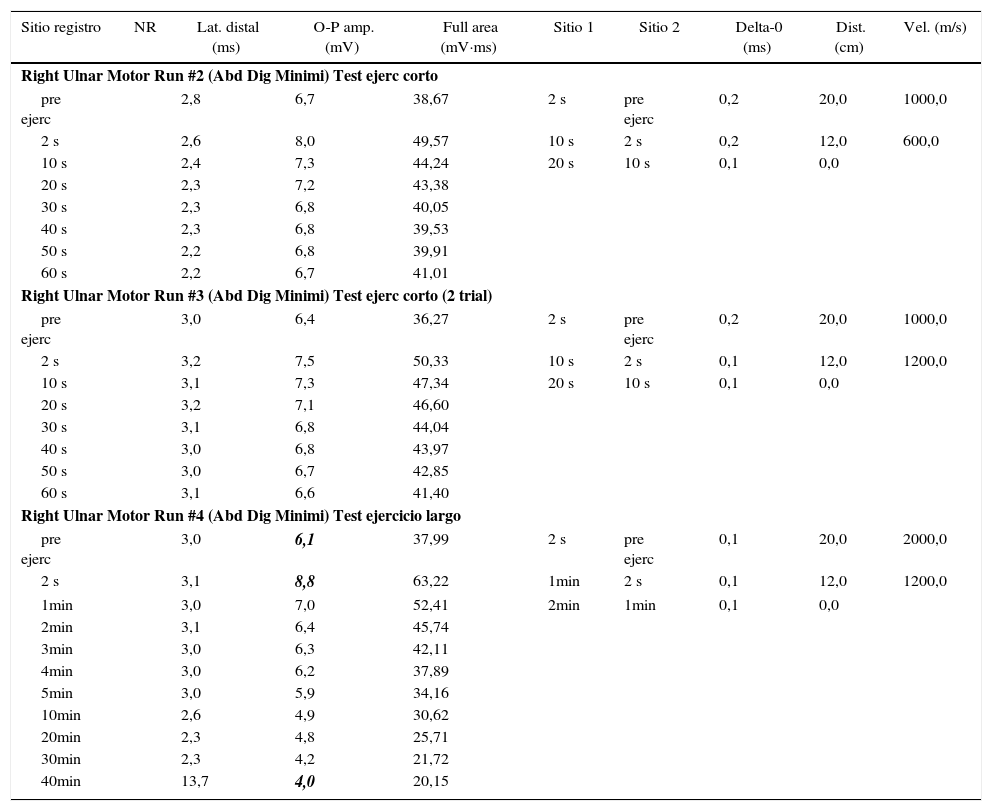
Se realizó un electromiograma convencional con estudio de velocidades de conducción y estimulación repetitiva, el cual resultó normal. Se efectuó posteriormente protocolo de estudio electrofisiológico según Fournier et al. El test corto no arrojó resultados relevantes. En el test largo se evidenció un aumento significativo del potencial de acción muscular compuesto (CMAP) postejercicio (+32%) con posterior caída (–34%), que se hizo evidente a los 10 min de iniciado el test largo, determinando un patrón tipo iv (tabla 1). Se decidió, en consecuencia, realizar la secuenciación genética de CACNA1S, sin encontrar hallazgos patológicos, y de SCN4A, encontrando una variante en heterocigosis en el exón 5, c.664C>G; p.R222G (fig. 2). Dicha variante, no descripta previamente, es predictiva de ser patogénica (herramientas informáticas Mutation Taster [probably damaging], Polyphen2 [damaging], SIFT y PROVEAN [deleterious]), además de haberse reportado 2 casos previamente, con otro cambio aminoacídico en la misma posición (p.R222W)7. Se llegó de esta manera al diagnóstico de certeza de parálisis periódica hipocalémica tipo ii.
Protocolo electrofisiológico del paciente según Fournier et al
| Sitio registro | NR | Lat. distal (ms) | O-P amp. (mV) | Full area (mV·ms) | Sitio 1 | Sitio 2 | Delta-0 (ms) | Dist. (cm) | Vel. (m/s) |
|---|---|---|---|---|---|---|---|---|---|
| Right Ulnar Motor Run #2 (Abd Dig Minimi) Test ejerc corto | |||||||||
| pre ejerc | 2,8 | 6,7 | 38,67 | 2 s | pre ejerc | 0,2 | 20,0 | 1000,0 | |
| 2 s | 2,6 | 8,0 | 49,57 | 10 s | 2 s | 0,2 | 12,0 | 600,0 | |
| 10 s | 2,4 | 7,3 | 44,24 | 20 s | 10 s | 0,1 | 0,0 | ||
| 20 s | 2,3 | 7,2 | 43,38 | ||||||
| 30 s | 2,3 | 6,8 | 40,05 | ||||||
| 40 s | 2,3 | 6,8 | 39,53 | ||||||
| 50 s | 2,2 | 6,8 | 39,91 | ||||||
| 60 s | 2,2 | 6,7 | 41,01 | ||||||
| Right Ulnar Motor Run #3 (Abd Dig Minimi) Test ejerc corto (2 trial) | |||||||||
| pre ejerc | 3,0 | 6,4 | 36,27 | 2 s | pre ejerc | 0,2 | 20,0 | 1000,0 | |
| 2 s | 3,2 | 7,5 | 50,33 | 10 s | 2 s | 0,1 | 12,0 | 1200,0 | |
| 10 s | 3,1 | 7,3 | 47,34 | 20 s | 10 s | 0,1 | 0,0 | ||
| 20 s | 3,2 | 7,1 | 46,60 | ||||||
| 30 s | 3,1 | 6,8 | 44,04 | ||||||
| 40 s | 3,0 | 6,8 | 43,97 | ||||||
| 50 s | 3,0 | 6,7 | 42,85 | ||||||
| 60 s | 3,1 | 6,6 | 41,40 | ||||||
| Right Ulnar Motor Run #4 (Abd Dig Minimi) Test ejercicio largo | |||||||||
| pre ejerc | 3,0 | 6,1 | 37,99 | 2 s | pre ejerc | 0,1 | 20,0 | 2000,0 | |
| 2 s | 3,1 | 8,8 | 63,22 | 1min | 2 s | 0,1 | 12,0 | 1200,0 | |
| 1min | 3,0 | 7,0 | 52,41 | 2min | 1min | 0,1 | 0,0 | ||
| 2min | 3,1 | 6,4 | 45,74 | ||||||
| 3min | 3,0 | 6,3 | 42,11 | ||||||
| 4min | 3,0 | 6,2 | 37,89 | ||||||
| 5min | 3,0 | 5,9 | 34,16 | ||||||
| 10min | 2,6 | 4,9 | 30,62 | ||||||
| 20min | 2,3 | 4,8 | 25,71 | ||||||
| 30min | 2,3 | 4,2 | 21,72 | ||||||
| 40min | 13,7 | 4,0 | 20,15 | ||||||
Obsérvese que el test de ejercicio corto no mostró modificaciones significativas del potencial de acción compuesto (CMAP), mientras que en el test largo se evidenció un aumento significativo del CMAP postejercicio (+32%) con posterior caída (–34%), que se hizo evidente a los 10 min de iniciado el test largo, determinando un patrón tipo iv.
 Electroferograma de caso índice: se observa variante en heterocigosis en el exón 5, c.664C>G; p.R222G. B) Forma wild type.")
Las canalopatías son procesos patológicos caracterizados por alteraciones en los canales iónicos. Los canales iónicos son glicoproteínas transmembrana que median la excitabilidad celular. Pueden encontrarse en 3 estados: abiertos, cerrados (reposo) e inactivados (período refractario al paso de iones). Se conforman de subunidades, cada una de las cuales es codificada por un gen diferente. Tienen múltiples funciones, entre ellas, la generación del potencial de acción, la estabilización del potencial de membrana, la repolarización celular, la liberación de neurotransmisores, la contracción muscular, la señalización intracelular, la hiperpolarización de la célula, etc. Existen 2 grandes grupos de canales iónicos: los dependientes del voltaje y los dependientes del ligando. Los canales dependientes del voltaje son activados por cambios en el potencial de transmembrana. El poro del canal constituye la abertura por donde los iones atraviesan selectivamente la membrana. Los canales iónicos experimentan cambios conformacionales que provocan la apertura y el cierre del poro del canal en cuestión de milisegundos (estos cambios conformacionales se llaman colectivamente compuertas o gating). La apertura está acoplada a mecanismos que detectan señales celulares, como cambios en el voltaje, señales químicas (un neurotransmisor, niveles de calcio o adenosintrifosfato) o señales mecánicas. Cuando se abren, el movimiento de iones por su través genera las señales eléctricas de las neuronas y otras células excitables, como las musculares2-8.
Las parálisis periódicas hipopotasémicas son canalopatías iónicas dependientes del voltaje de etiología genética con patrón de herencia autosómico dominante. Secuenciaciones genéticas han demostrado que mutaciones en el gen que codifica la subunidad alfa 1 de los canales de calcio del músculo esquelético (CACNA1S) son la causa de la mayoría de las parálisis periódicas hipopotasémicas (parálisis periódica hipopotasémica tipo i (PPhipoK I)). Mutaciones en los genes que codifican la subunidad alfa 1 de los canales de sodio (SCN4A) son causa de un número menor de casos (parálisis periódica hipopotasémica tipo ii [PPhipoK II]). El KCNJ18 mutado también han sido implicado en un pequeño porcentaje de pacientes7-9.
La subunidad alfa 1 de estos canales está compuesta por 4 dominios homólogos (DI-DIV), cada uno de los cuales se forma por 6 segmentos transmembrana. Tres puntos de mutación que afectan a la arginina han sido encontrados en el segmento 4 (S4) de los dominios ii y iv (R528H, R1239H y R1239G) (el dominio iii ha sido también reportado, sobre todo en presentaciones clínicas más tempranas). Las mutaciones de R528H y R1239H son responsables de la mayoría de los casos de PPhipoK I y su incidencia relativa varía de una población a otra. Se ha observado una penetrancia incompleta en mujeres y un comienzo más tardío de la enfermedad en las mutaciones de R528H. El gen que codifica para SCN4A se encuentra en el cromosoma 17q22-23. Sus mutaciones afectan principalmente al DII del S4 de la subunidad alfa 19.
Estos cambios conformacionales anómalos, como se menciona previamente, afectan a los residuos de arginina de los dominios sensibles al voltaje, preferentemente DII y DVI, lo que provoca una pérdida de la función del canal, disminuyendo la densidad de los mismos y generando poros aberrantes de corriente iónica (aberrant «gating pore current»). Esto lleva a la activación del canal en el estado de reposo, con salida de cationes y acumulación de protones intracelulares, alteración de la homeostasis celular por cambios en el pH y despolarización paradójica de la membrana, produciendo una menor densidad de corriente iónica, que se traduce en menor excitabilidad muscular y clínicamente en parálisis motora10.
Se sabe que el ejercicio físico en estos pacientes actúa como desencadenante, agravante o atenuante de los síntomas, según las diferentes formas patológicas. Basado en esto, el ejercicio puede ser utilizado como un test funcional en electromiografía para mejorar la sensibilidad y lograr categorizaciones diagnósticas aproximadas. El protocolo diseñado por Fournier et al. mide el CMAP en las pruebas electrofisiológicas de conducción nerviosa sobre 2 tipos de test que implican ejercicio físico: un test corto y un test largo, que se diferencian mínimamente en lo procedimental, requieren diferentes tiempos, utilizan el mismo concepto fisiopatológico y su información se analiza en forma complementaria (tabla 2). Los resultados pueden determinar patrones patológicos que nos ayudan a estimar (en contextualización con las características clínicas del paciente) el tipo de canalopatía, los genes implicados y, de manera indirecta, el éxito o fracaso terapéutico de los fármacos a utilizar6.
Descripción básica de protocolo según Fournier et al
| Protocolo de Fournier et al. |
|---|
| 1. Mantener temperatura entre 32-34°C |
| 2. Antes del ejercicio, realizar estímulos repetidos cada 10 s por 2 min para ver estabilidad del CMAP |
| 3. Realizar estimulaciones repetitivas a 3Hz por 10 repeticiones, luego 1 min de descanso previo inicio de test del ejercicio |
| Prueba de ejercicio corto: contracción muscular durante 10 s; luego registro a los 2-10-20-30-40-50-60 s. Realizar 3 repeticiones del test |
| Prueba del ejercicio largo: 5 min de contracción muscular con 3-4 s de descanso cada 30 s. Posteriormente, registro a los 2 s y luego cada 1 min durante 5 min y cada 5 min durante 40 min |
El inhibidor de la anhidrasa carbónica, acetazolamida, ha sido usado durante décadas como tratamiento profiláctico para reducir la severidad y la frecuencia de ataques. Sin embargo, solo el 50% de los pacientes tienen respuesta favorable, pudiendo ocurrir efectos adversos que limiten su uso en algunos pacientes y en otros, incluso, empeorar los ataques de parálisis. Esto ocurre sobre todo en PPhipoK II3.
Se sabe que la despolarización paradójica que precipita los ataques no solo es debida al flujo de corriente iónico anómalo producto de los poros aberrantes, sino que también depende del gradiente clorhídrico transmembrana, ya que altas concentraciones de cloro intramuscular promueven la despolarización en condiciones de hipopotasemia. La acumulación clorhídrica en el músculo es derivada del cotransportador sodio-potasio-2cloro (ctNKCC), el cual facilita el influjo de iones. Este ctNKCC es potencialmente inhibido por el diurético bumetanida. En un modelo animal mutante (NaV1.4-R669H) se observó que el uso de este fármaco fue capaz de prevenir la debilidad muscular4,5 y, según los autores, podría considerarse como alternativa terapéutica farmacológica en aquellos pacientes que sean diagnosticados de PPhipoK II y en cualquiera de las variedades de PPhipoK que respondan inadecuadamente con los tratamientos convencionales. En conclusión, podemos destacar, sobre la base de lo expuesto, que el conocimiento clínico, la caracterización electrofisiológica y la secuenciación molecular de estos cuadros patológicos permiten el diagnóstico preciso y orientan a la elección farmacológica adecuada.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesNo se declaran conflictos de intereses.
