




Las encefalopatías epilépticas del lactante, de origen genético, se manifiestan en los 2 primeros años de vida. La particularidad de este cuadro es la refractariedad al tratamiento con fármacos antiepilépticos habituales y la presencia de neuroimágenes, estudios metabólicos y cariotipos convencionales normales.
Caso clínicoNuestro objetivo es la descripción de una paciente de 5 años con diagnóstico de encefalopatía epiléptica infantil temprana (EEIT) tipo 42, asociada a mutación de novo del canal de calcio CACNA1A. En la misma determinación, presentó mutación del canal de sodio SCN5A asociado a síndrome de Brugada.
ConclusiónComo conclusión, la interacción entre ambos síndromes genéticos complejizó la decisión farmacológica.
Epileptic encephalopathies in infants, of genetic origin, manifest in the first two years of life. The peculiarity of this picture is the refractoriness to treatment with usual antiepileptic drugs and the presence of normal neuroimaging, metabolic studies and conventional karyotypes.
Clinical caseOur objective is the description of a 5-year-old patient diagnosed with early childhood epileptic encephalopathy type 42, associated with a de novo mutation of the calcium channel CACNA1A. In the same determination, the patient presented a mutation of the sodium channel SCNSA associated with Brugada syndrome.
ConclusionIn conclusion, the interaction between both genetic syndromes complicated the pharmacological decision.
La definición de encefalopatía epiléptica (EE) de la Liga Internacional contra la Epilepsia (ILAE) adjudica este término a la actividad epileptiforme que por sí contribuye a generar alteración en el desarrollo y cambios en el comportamiento severos más allá de lo esperado por la patología subyacente por sí misma (por ejemplo, malformaciones del desarrollo cortical). Implican un desarrollo más lento o bien retraso en el desarrollo preexistente y, usualmente, inician en la infancia y la niñez1. Las EE se producen no solo por la acción de actividad epileptiforme difusa y frecuente en el electroencefalograma (EEG), sino también por la repetición de crisis en pacientes que pueden tener un EEG normal interictal en las primeras etapas de la enfermedad2. Es importante determinar la etiología específica de cada entidad debido a que puede estar asociada a la afectación de otros órganos y se deben implementar tratamientos en conjunto que no resulten nocivos para las patologías en cuestión. Siempre debe descartarse en primera instancia aquellas causas que sean plausibles de tratamiento específico (infecciones, enfermedades inflamatorias o autoinmunes del sistema nervioso central). En general, las convulsiones no son la única manifestación de estas entidades, sino que también se asocian con un fenotipo característico y otras alteraciones sistémicas asociadas.
La EEIT tipo 42 corresponde a una entidad neurológica caracterizada por el inicio de varios tipos de crisis epilépticas dentro de las primeras horas o días de vida, aunque algunos pacientes pueden tener comienzo de la clínica en las primeras semanas. Se hereda de forma autosómica dominante o bien mediante mutaciones de novo del gen CACNA1A ubicado en el brazo corto del cromosoma 19, que codifica la principal subunidad formadora de poros del canal de calcio tipo P/Q, alfa-1 (Cav2.1). Las crisis suelen ser refractarias al tratamiento convencional y están asociadas con anormalidades electroencefalográficas que incluyen espigas multifocales y complejos onda-espiga generalizados. Aparecen usualmente en la niñez presentando, además de alteración en el desarrollo cognitivo y cambios en el comportamiento, hipotonía de músculos axiales, hipertonía periférica con hiperreflexia, temblor, ataxia y movimientos oculares anormales3. El tratamiento de primera línea es lamotrigina, bloqueante de canales de sodio.
El síndrome de Brugada es una arritmia cardíaca hereditaria (autosómica dominante, penetrancia incompleta) caracterizado por hallazgos en el electrocardiograma (ECG) característicos, no relacionado con isquemia, alteraciones electrolíticas o enfermedad cardíaca estructural evidente. Clínicamente puede manifestarse como un cuadro sincopal e, incluso, puede ser causante de muerte súbita. Varios fármacos están contraindicados en pacientes con esta afección, entre ellos lamotrigina.
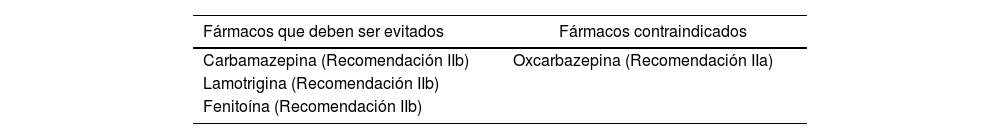
Nuestro caso clínico trata de un paciente que presenta diagnóstico de EEIT tipo 42 cuyo tratamiento de primera línea es lamotrigina, con asociación a síndrome de Brugada, en el cual se contraindica (así como también fenitoína, carbamazepina y oxcarbazepina) (tabla 1)4.
Fármacos antiepilépticos contraindicados en el síndrome de Brugada
| Fármacos que deben ser evitados | Fármacos contraindicados |
|---|---|
| Carbamazepina (Recomendación IIb) | Oxcarbazepina (Recomendación IIa) |
| Lamotrigina (Recomendación IIb) | |
| Fenitoína (Recomendación IIb) |
Fuente: Postrema et al., 20094.
Paciente de 5 años de edad con diagnóstico de EEIT tipo 42 y síndrome de Brugada, sin antecedentes familiares de relevancia. Su protocolo de vacunación era completo. Fue recién nacida de término con peso adecuado para su edad gestacional, con embarazo controlado y serologías maternas negativas. Requirió internación por 10 días en Neonatología, por convulsión tónico-clónica generalizada el primer día de vida. Se realizó ecografía cerebral, abdominal y EEG dentro de parámetros normales. Continuó en tratamiento con fenobarbital hasta los 6 meses.
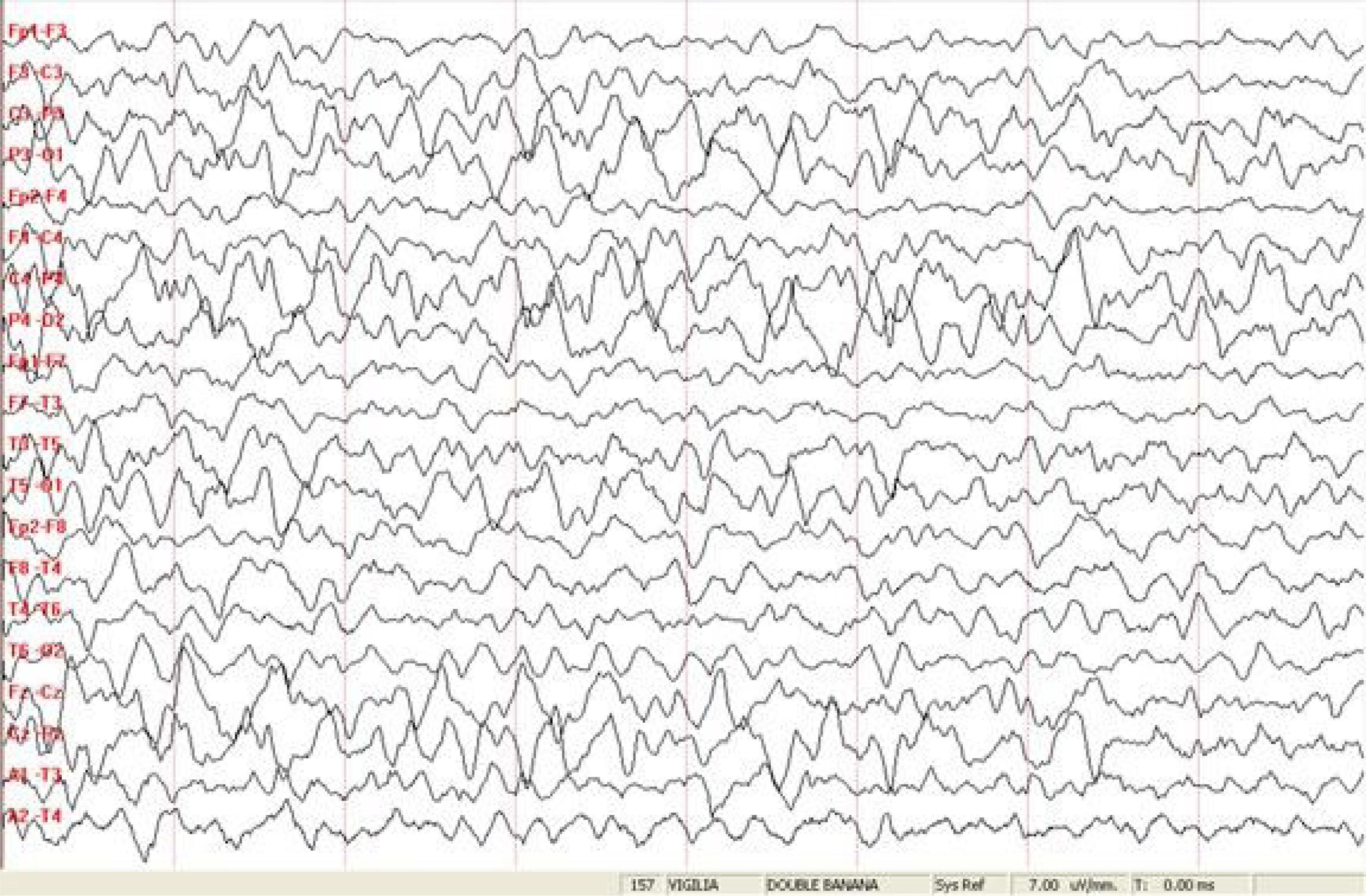
A los 9 y 10 meses requirió internación en Unidad de Terapia Intensiva Pediátrica (UTIP) por estatus epiléptico (SE). En la segunda oportunidad, tuvo requerimiento de asistencia respiratoria mecánica (ARM). Los estudios complementarios de EEG y RMN de cerebro fueron normales. Se agregó levetiracetam (LVT) al tratamiento de base. Presentó múltiples intercurrencias infectológicas, con cuadros de vías aéreas superiores en distintas ocasiones por Rinovirus, Adenovirus y COVID 19 con descompensación de su patología de base. Las crisis cedieron con carga de benzodiacepinas y tuvo requerimiento de ARM en dos oportunidades. El EEG mostró disfunción frontal bilateral de predominio izquierdo dado por ondas lentas y ondas agudas en el hemisferio izquierdo. En la última internación en julio de 2021 se ajustó la medicación de base fenobarbital 5mg/kg/día, ácido valproico (AVP) 50mg/kg/día, LVT 70mg/kg/día y clobazam 5mg/día. Permaneció sin crisis en los últimos 6 meses.
Con respecto al estudio genético, presentó alteración en el gen CACNA1A (brazo corto del cromosoma 19) correspondiente a alteración en los canales de calcio voltaje dependientes de tejido neuronal, asociada a síndromes neurológicos: encefalopatía epiléptica EEIT 42, ataxia espinocerebelosa tipo 6, ataxia episódica tipo 2 y migraña familiar hemipléjica 1-. También se detectó una alteración en el gen SCN5A, en el brazo corto del cromosoma 3, correspondiente a alteración en los canales de sodio voltaje dependientes de tejido miocárdico asociado a síndrome de Brugada.
Actualmente, presenta retraso global del desarrollo, concurre a escolaridad común, en Sala de 5 años, con maestra integradora, realiza terapias de apoyo con Psicopedagogía y estimulación temprana en forma semanal. Continúa en seguimiento por Neurología y Cardiología Infantil.
ComentariosNuestro caso clínico trata de un paciente sin antecedentes personales ni familiares de relevancia que presenta diagnóstico de EEIT tipo 42 con asociación a síndrome de Brugada, ambos detectados mediante estudio genético (análisis molecular del exoma) siendo estas mutaciones de novo. Requirió múltiples internaciones debido a que las crisis epilépticas eran refractarias al tratamiento, algunas de ellas con requerimiento de ARM. Actualmente, se encuentra en tratamiento combinado con varios fármacos manteniéndose libre de crisis.
En comparación con la bibliografía actual, se sabe que la genética juega un papel importante en muchos síndromes epilépticos; sin embargo, se han descubierto genes específicos solo en una pequeña proporción de casos. Los estudios de asociación del genoma para epilepsias focales y generalizadas han revelado pocas asociaciones significativas, y las variantes raras del número de copias explican solo un pequeño porcentaje de los casos3. El diagnóstico se realiza mediante análisis molecular de exoma clínico dirigido a variantes puntuales (SNPs) y deleciones/inserciones en regiones exónicas e intrónicas adyacentes, así como variantes en el número de copias (CNVs) en genes candidatos asociados a epilepsias infantiles.
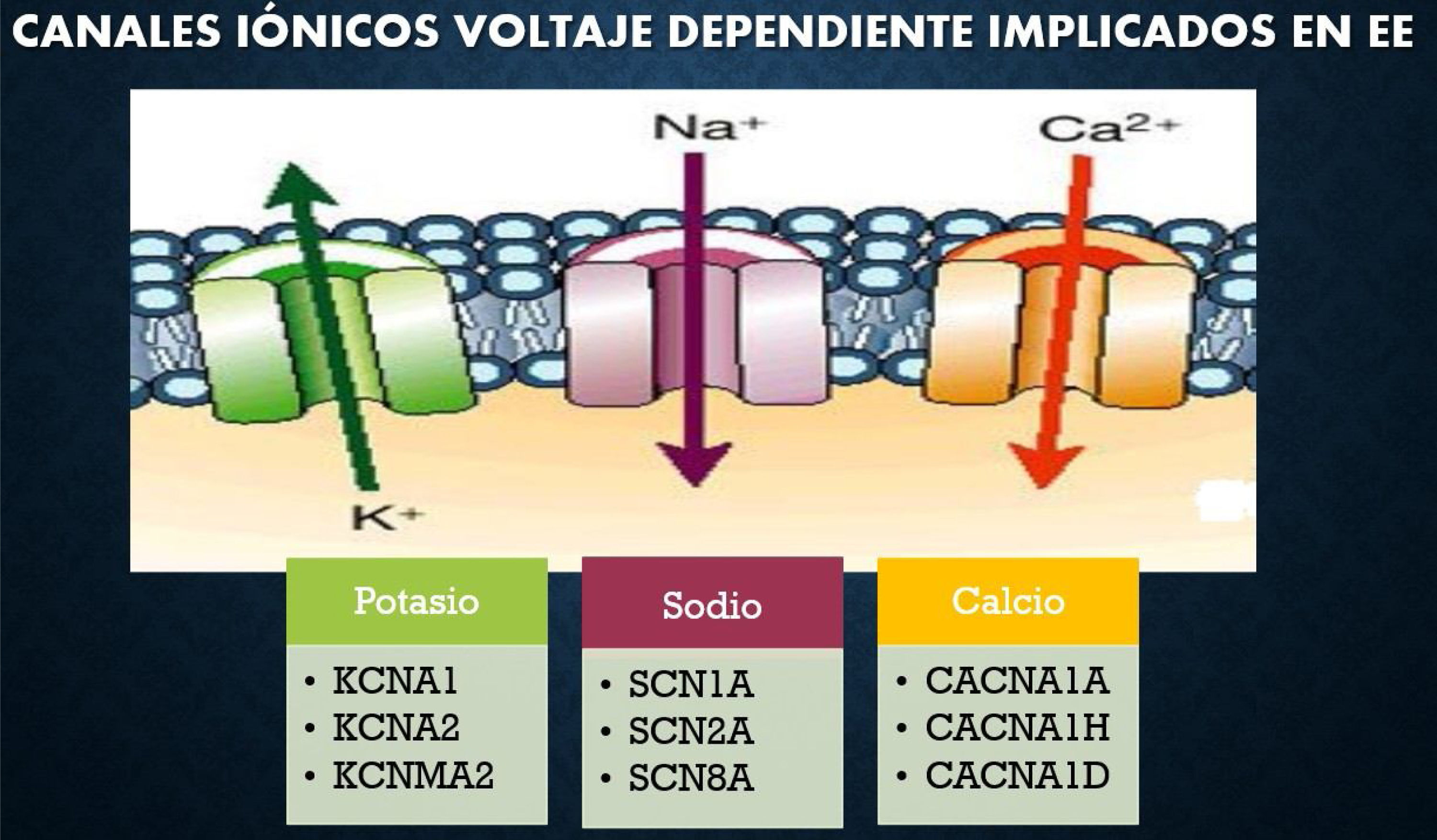
Un paradigma emergente en los trastornos neuropsiquiátricos es el gran impacto de las mutaciones de novo en el riesgo de desarrollar enfermedad. Se definen como encefalopatías epilépticas un grupo heterogéneo de trastornos epilépticos graves, caracterizados por la aparición temprana de convulsiones, con afectación cognitiva y conductual asociada con la actividad epiléptica. Los genes SCN1A, STXBP1, SCN8A, SCN2A, CACNA1A y CDKL5 estarían relacionados con EE (fig. 1)3.
.")
El gen CACNA1A, ubicado en el cromosoma 19p13, codifica la principal subunidad formadora de poros del canal de calcio tipo P/Q, alfa-1 (conocido como Cav2.1). Este canal desempeña un papel fundamental en el acoplamiento de la entrada de calcio y se produce la exocitosis vesicular. Esta reacción media la entrada de este ion, lo que induce la despolarización en dendritas, cuerpos celulares y terminales nerviosas (fig. 2)5.
.")
Ejemplos de canales iónicos implicados en EE3.
Fuente: Modificado de Epi4K and EPGP Investigators. (2013).
Los resultados de un estudio de cohorte publicado en 2016 por EpiK4 Consortium demostraron evidencia definitiva que las mutaciones de novo en SLC1A2 y CACNA1A también eran causa de EE. La cohorte comprendía 531 personas con diagnóstico de EE de causa desconocida, sin mutaciones en genes asociados a epilepsia ya conocidos. Se descubrieron variantes patógenas o probablemente patógenas en siete de los 27 genes que fueron secuenciados (ALG13, CACNA1A, DNM1, GABRB3, GNAO1, IQSEC2 y SLC1A2). Las mutaciones en los genes GABRB3 y CACNA1A fueron las contabilizadas en la mayor parte de individuos. Se identificaron mutaciones patogénicas en CACNA1A en cuatro de los pacientes y un hermano afectado. Este número de mutaciones sin sentido de novo para esta cohorte representó evidencia clara de las alteraciones del canal CACNA1A como causa de EEIT6.
Por otra parte, el síndrome de Brugada es una arritmia cardíaca hereditaria (autosómica dominante, penetrancia incompleta) caracterizado por la elevación del ST en forma de curva y la elevación del punto J de al menos 2mm en al menos dos de las derivaciones precordiales derechas (V1-V3) en el ECG no relacionado con isquemia, alteraciones electrolíticas o enfermedad cardíaca estructural evidente. Clínicamente puede manifestarse como un cuadro sincopal e, incluso, puede ser causante de muerte súbita7. El diagnóstico requiere la presencia del patrón característico en el ECG y al menos uno de los siguientes criterios clínicos reconocidos: síncope, paro cardíaco previo (documentado o taquicardia ventricular polimórfica inducible o fibrilación ventricular), antecedentes familiares de muerte súbita antes de los 45 años o patrón del ECG y/o respiración agónica nocturna7.
La mayoría de los casos con síndrome de Brugada son causados por mutaciones en el gen del canal de sodio SCN5A que codifica la subunidad alfa. Recientemente, se han descrito nuevas mutaciones sin sentido con pérdida de función en CACNA1C (A39V y G490R) y CACNB2 (S481L) que codifican las subunidades alfa1 y beta 2b del canal de calcio tipo L que también podrían asociarse a este síndrome. Fármacos como antidepresivos tricíclicos, litio, antihistamínicos de primera generación y cocaína son medicamentos que presentan el síndrome de Brugada como efecto adverso7. Fármacos antiarrítmicos de clase I, incluidos flecainida, propafenona y procainamida inhiben el comienzo del potencial de acción cardíaco mediante el bloqueo de canales de sodio, razón por la cual constituyen una contraindicación absoluta para los pacientes con síndrome de Brugada. Para el caso particular de fármacos antiepilépticos, estarían contraindicados aquellos que comparten el mecanismo de acción mencionado para los antiarrítmicos: por ejemplo, lamotrigina, que en dosis elevadas puede perder la especificidad sobre receptores neuronales y generar cambios en la contracción cardíaca al actuar sobre canales de Sodio del miocardio y actuaría como un disparador potencial del síndrome de Brugada8,9 (tabla 1).
El reporte de un caso publicado en 2016 por Byers et al. describe la evolución con el inicio de lamotrigina de una paciente con diagnóstico de EEIT tipo 42 asociada a mutación del canal CACNA1C luego del inicio del tratamiento con zonisamida y AVP. Aunque se pensaba que actuaba en los canales de sodio, la lamotrigina también modula la actividad del canal de calcio tipo P/Q, lo que la convierte en candidata de primera línea para una terapia de precisión para pacientes con EE por variantes patogénicas CACNA1A5.
ConclusiónPara el caso de la paciente, la combinación de ambos síndromes resultó ser una gran controversia a la hora de determinar el tratamiento adecuado para el control de las convulsiones, sin afectar la función cardíaca. Se probaron varias opciones de tratamiento en diferentes intercurrencias; logró permanecer sin crisis con fenobarbital, LVT, AVP, y clobazam a dosis adecuadas a la edad y al peso de la paciente. El estudio genético, no siempre disponible en etapas tempranas de la vida, permitió el tratamiento adecuado y el control de las crisis, sin poner en riesgo la vida de la niña.
Si bien es un caso aislado, se logró el control de las crisis por períodos prolongados. Llama la atención, la imposibilidad de suspender el fenobarbital a pesar del intento de rotación aún con dosis óptimas con el resto del esquema farmacológico.
Conflicto de interesesNo se recibió ningún tipo de financiación ni se presentan conflictos de interés para la realización de este artículo.
