




Las paraplejías espásticas hereditarias complicadas (PEHC) están caracterizadas por presentar síntomas neurológicos acompañantes de la lesión característica de los haces corticoespinales. En las formas asociadas a atrofia de cuerpo calloso, las mutaciones más frecuentes son aquellas vinculadas a SPG11 (espastacsina) y en menor medida a SPG15.
Caso clínicoPaciente de 39 años de origen italiano, desde los 23 años desarrolla inestabilidad de la marcha, espasticidad progresiva en piernas y caídas frecuentes sin trastornos esfinterianos. Dicha sintomatología paulatinamente fue acentuándose hasta el punto de incapacitar completamente su deambulación. Los estudios genéticos detectaron 2 mutaciones heterocigotas compuestas en SPG11.
ConclusionesSe describen los hallazgos típicos en la paraplejía espástica hereditaria complicada asociada a atrofia del cuerpo calloso por 2 nuevas mutaciones en SPG11, no reportadas previamente. Ambas variantes son predictivas de ser patogénicas y segregan con la enfermedad en la familia de la paciente. La presencia de atrofia de cuerpo calloso en el contexto de un cuadro de paraplejía espástica hereditaria complicada, sea como signo temprano o tardío, indica en primera instancia mutaciones en SPG11.
Complicated hereditary spastic paraplegias (CHSP) embrace conditions in which different neurological symptoms are present other than the «classical» corticospinal involvement. In cases associated with corpus callosum atrophy, mutations in SPG11 and less frequently in SPG15, are common findings.
Case report39 year-old Italian patient with normal birth and development, that began around 23 years of age with gait problems defined as progressive instability, stiffness in her legs and falls. There was no sphincter involvement. These conditions slowly progressed up to the point to impede her walking. Genetic investigations detected two novel compound heterozygous mutations in SPG11.
ConclusionsWe describe typical clinical findings in a case with complicated hereditary spastic paraplegia associated with corpus callosum atrophy due to two novel mutations in SGP11. Both variants are predicted to be pathogenic and segregate with disease in her family. Corpus callosum atrophy either as early or late finding should prompt SPG11 mutation analysis.
En 1875 el médico alemán Wilhelm Heinrich Erb en su ensayo Spastische Spinalparalyse realiza la primera descripción clínica de un caso de paraplejía espástica hereditaria (PEH)1. Cinco años más tarde, Adolf Strümpell reporta de forma más sistematizada numerosos casos, realizando aportes significativos en relación con los hallazgos anatomopatológicos y del compromiso predominante de los haces corticoespinales2. Años después, el neurólogo francés Maurice Lorrain profundiza las descripciones clínicas iniciadas previamente por el médico alemán. Por dichos valiosos aportes, la PEH es también acuñada como enfermedad de Strümpell-Lorrain.
La PEH incluye un grupo de condiciones en las cuales existe como hallazgo anatomopatológico preponderante la degeneración retrógrada de los tractos corticoespinales y las columnas posteriores de la médula espinal. Contrariamente a esta aparente «uniformidad» clínica, genotípicamente difiere en sus formas de transmisión (autosómica dominante, recesiva y ligada al X recesiva), en sus mecanismos fisiopatogénicos y en la asociación genética con más de 52 loci diferentes.
El término PHC complicada (PEHC) se reserva para los casos en los cuales, además del compromiso corticoespinal, existen otros síntomas como epilepsia, deterioro cognitivo, ataxia, amiotrofia severa, neuropatía y atrofia óptica, entre otros3. La presencia de atrofia del cuerpo calloso (ACC) en la paraplejía espástica complicada ha demostrado ser predictiva de mutaciones en genes específicos como SPG11 o SPG154-6.
Este reporte describe los hallazgos clínicos, moleculares, imagenológicos y electrofisiológicos en una paciente con PEHC asociada a ACC producida por 2 nuevas mutaciones en SPG11.
Caso clínicoPaciente de 39 años, con antecedentes perinatológicos y del desarrollo normales. A los 23 años desarrolla inestabilidad en la marcha, espasticidad en los miembros inferiores de carácter progresivo y caídas frecuentes sin trastornos esfinterianos. Dicha sintomatología fue acentuándose paulatinamente hasta incapacitar completamente su deambulación.
Sus padres son sanos y presentan antecedentes de consanguinidad familiar (abuelos paternos primos en primer grado), ambos de origen italiano. Dos hermanas, una menor, afectada con similares síntomas. En estos últimos 2 años, la paciente relata la aparición de debilidad y atrofia muscular distal y simétrica en los miembros superiores, que motiva la realización de un electromiograma.
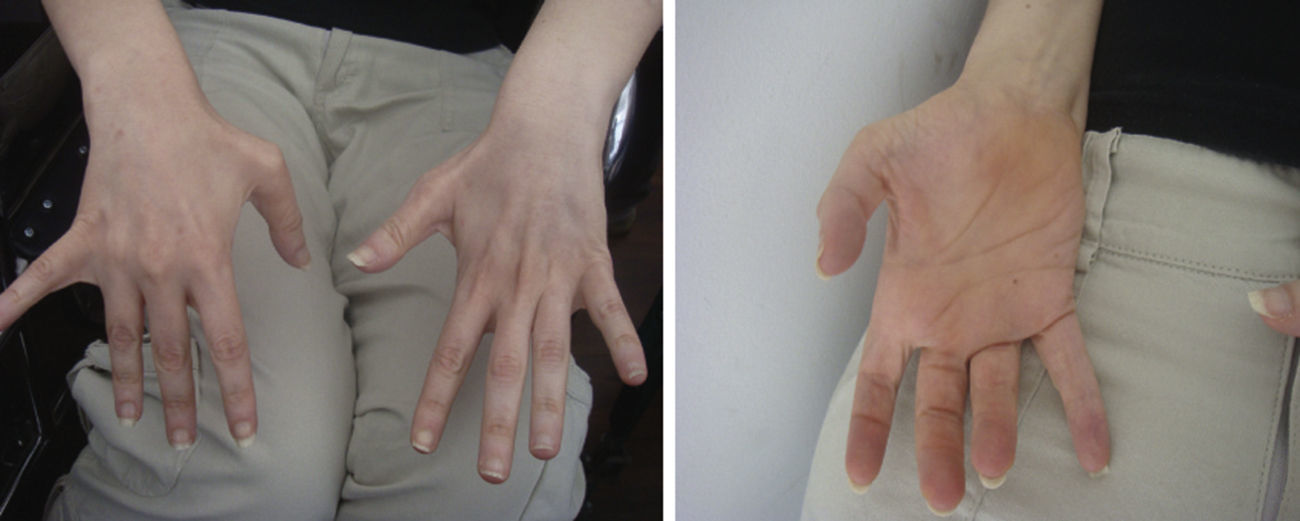
Examen neurológicoLucida, Mini Mental State Evaluation 22/30, fondo de ojo normal sin retinitis pigmentaria, inestabilidad en la fijación visual, sin trastorno deglutorio, voz escandida. No presenta atrofia lingual ni fasciculaciones. En los miembros superiores, presenta hiperreflexia, tono conservado, taxia relativamente conservada, atrofia y debilidad distal simétrica de músculos intrínsecos de las manos (fig. 1). En los miembros inferiores, presenta espasticidad severa, reflejos policinéticos, reflejo plantar flexor, clonus agotable, debilidad muscular distal, sin presencia de pie cavo. Leve hipopalestesia distal, taxia no evaluable debido a la severa hipertonía.
Estudio complementarios
Los estudios de laboratorio fueron normales, incluyendo ácidos grasos de cadena muy larga, arilsulfatasa A, creatincinasa y aldolasa. Virus de leucemia/linfoma de células T del ser humano (HTLV 1-2), negativos.
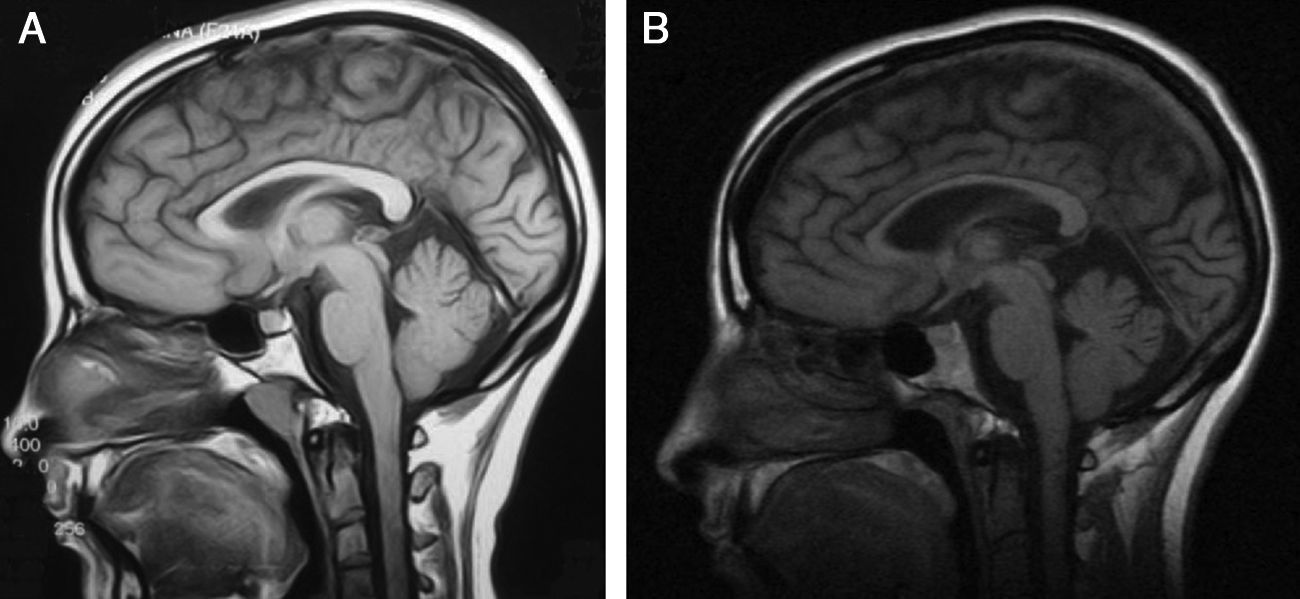
Electromiograma (año 2000): neuropatía periférica de tipo axonal en ambos miembros inferiores. Electrocardiograma: normal. Biopsia de músculo: agrupamiento histoquímico y compromiso de neurona motora inferior. Microscopia electrónica: aisladas fibras con leve incremento de la actividad oxidativa mitocondrial. Una resonancia magnética (RM) cerebral 15 años antes de su evaluación actual mostró como única anormalidad cuerpo calloso ligeramente adelgazado. Su RM actualmente muestra adelgazamiento marcado de cuerpo calloso, atrofia cortical difusa leve, hiperintensidad periventicular. RM de columna cervical y dorsal, normales (fig. 2).
 Año 1998. Se informó un incipiente adelgazamiento del cuerpo calloso. B) Año 2013. En la misma secuencia y corte, se observa progresión marcada y notable de la atrofia del cuerpo calloso.")
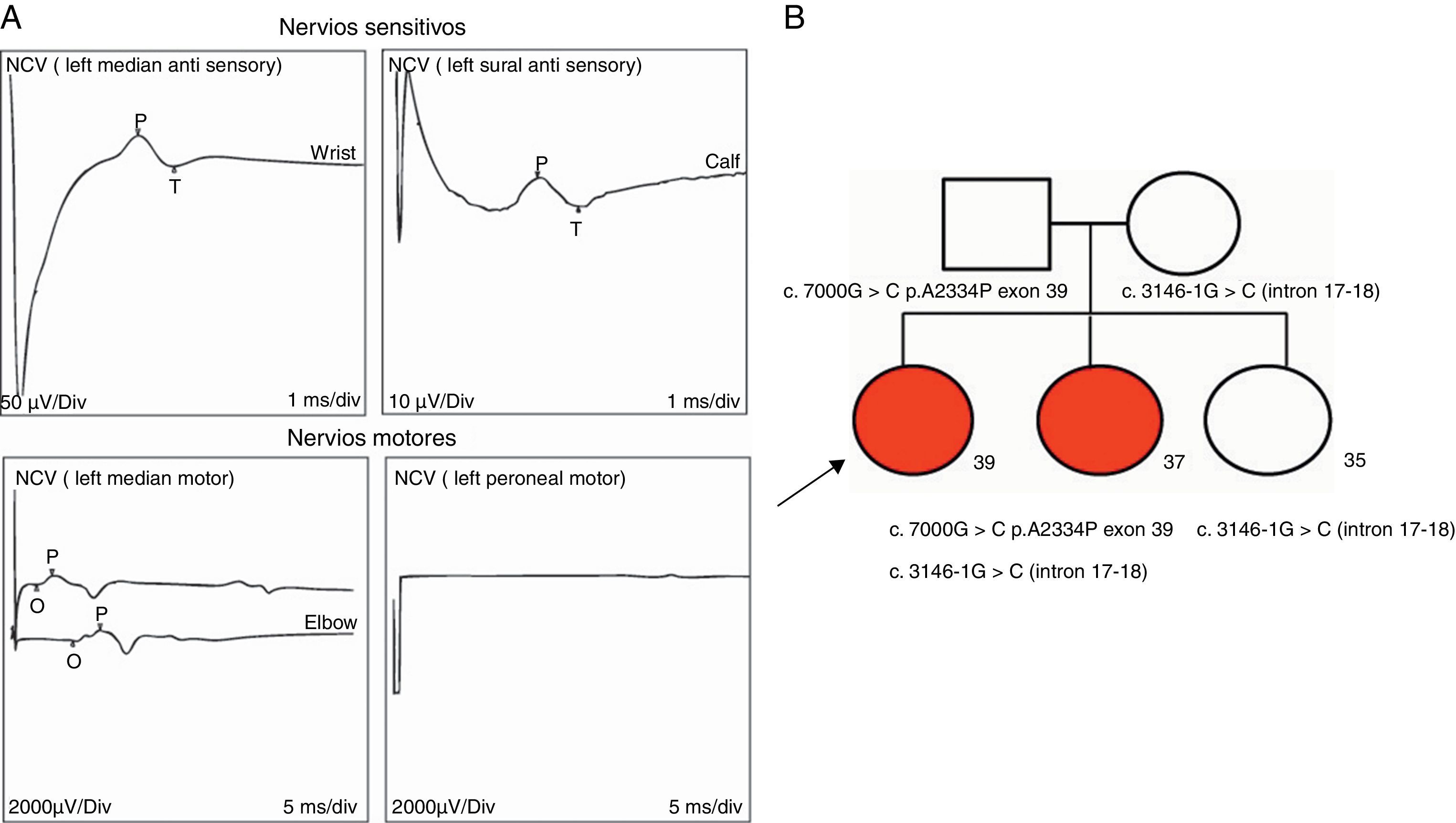
Electromiograma (año 2012): velocidades y amplitudes sensitivas normales, velocidades motoras normales con reducción de amplitudes. Los potenciales de unidad motora se hallaron severamente reducidos en su reclutamiento, con amplitudes muy incrementadas (> 15mV/div). Compatible con compromiso de motoneurona inferior/neuropatía axonal motora (fig. 3 A).
 Registros ilustrativos de los hallazgos electrofisiológicos compatibles con una neuropatía motora axonal/compromiso motoneurona inferior. Se observa degeneración de fibras motoras con indemnidad de amplitudes sensitivas. Se observaron potenciales de unidad motora (PUM) con marcado aumento de amplitud (15mV/div) (no mostrado). B) Pedigrí y segregación familiar de variantes halladas. Cuadrados indican varones, círculos mujeres. Formas rellenas, los miembros afectados. Las edades actuales están representadas con números debajo de formas geométricas..")
A) Registros ilustrativos de los hallazgos electrofisiológicos compatibles con una neuropatía motora axonal/compromiso motoneurona inferior. Se observa degeneración de fibras motoras con indemnidad de amplitudes sensitivas. Se observaron potenciales de unidad motora (PUM) con marcado aumento de amplitud (15mV/div) (no mostrado). B) Pedigrí y segregación familiar de variantes halladas. Cuadrados indican varones, círculos mujeres. Formas rellenas, los miembros afectados. Las edades actuales están representadas con números debajo de formas geométricas..
Desde el punto de vista molecular, en primera instancia se descartaron deleciones e inserciones en SPG11 mediante técnica molecular de multiple ligation probe amplification (en inglés). Posteriormente, se realizó la secuenciación completa de regiones exónicas e intrónicas adyacentes del gen SPG11 por metodología de Sanger. Se observaron 2 nuevas variantes en SPG11: c.7000 G>C, p.A2334P (exón 39) c.3146-1G>C (intrón17-18) trascrito ref. SPG11-001 (ENST00000261866, CCDS10112, NM_025137.3), presentes en heterocigosis en ambas hermanas afectadas, en tanto que la hermana restante no afectada era portadora solamente de la variante c.3146-1G>C (fig. 3 B). Ambas variantes segregaban familiarmente y no fueron descritas previamente como variantes polimórficas en la población general ni en pacientes previos. La predicción de patogenicidad de las mismas se realizó mediante técnicas de análisis informático (Human splicing finder, http://www.umd.be/HSF/; Polyphen2 http://genetics.bwh.harvard.edu/pph2/, y SIFT http://sift.jcvi.org/)7-9. La variante missense produce el cambio aminoacídico alanina a prolina, probablemente alterando la estructura tridimensional de la proteína, en tanto que la variante intrónica se localiza en el sitio de empalme con el exón 18, con probable impacto funcional. El software Polyphen calificó a la variante c.7000 G>C, p.A2334P como «probably damaging» con un score de 1,00 y el software SIFT como «damaging» con un score de 0,01. El software Human splicing finder calificó la variante c.3146-1G>C como «site broken».
ComentariosLas PEH incluyen un grupo heterogéneo de enfermedades neurodegenerativas que comparten entre sí la presencia de espasticidad progresiva en los miembros inferiores. La tradicional clasificación clínica establecía formas «puras», donde el compromiso parecería restringirse al tracto corticoespinal, y formas «complicadas», en donde diferentes síntomas neurológicos (p. ej., epilepsia, retraso mental, atrofia óptica, parkinsonismo) se combinan de forma variable con la manifestación cardinal de la paraparesia espástica.
La caracterización genética de estas enfermedades ha dado paso al descubrimiento de más de 52 loci diferentes (SPG1-52), en los cuales se describen formas recesivas, dominantes y ligadas al X, reflejando la heterogeneidad genotípica, así como diferentes mecanismos fisiopatogénicos y vías moleculares implicadas.
En las formas «puras», la forma de trasmisión más frecuente es de tipo autosómica dominante asociada, en alrededor de un 50%, a mutaciones en SGP4 y SPG3A10.
En contrapartida, las formas «complicadas» son subtipos menos frecuentes y habitualmente de carácter recesivo. En un tercio de estos casos la ACC puede estar presente y su presencia en estos casos es altamente indicativa de mutaciones en SPG1111.
La caracterización genética de la PEHC es ardua dadas la heterogeneidad genética, la extensión de los genes involucrados y la localización de las mutaciones, muchas de ellas en sitios de splicing de ubicación intrónica. En ciertas situaciones, la combinación de diferentes elementos clínicos de aparición temprana permite seleccionar el gen candidato; sin embargo, muchas de estas manifestaciones son de aparición solapada y tardía. Por este motivo, las técnicas de secuenciación masiva de última generación podrían constituir un método de caracterización genotípica más ágil, sobre todo en las formas recesivas12.
Este caso describe algunos de los hallazgos típicos presentes en la PEHC asociada a ACC (PEHC-ACC), en donde, además del compromiso corticoespinal, la presencia de deterioro cognitivo y atrofia muscular distal en los miembros superiores son hallazgos habituales.
En las formas PEHC-ACC, más del 52% de los casos están relacionados a mutaciones en SPG11. En un porcentaje menor de pacientes, SPG15 puede constituir el diagnóstico. Desde el punto de vista clínico, la mayoría de los pacientes presenta el inicio de sus síntomas alrededor de la segunda década de vida, su curso evolutivo es relativamente lento, en el cual más del 40% los pacientes pierden la deambulación de forma permanente alrededor de los 10 años de inicio de los primeros síntomas13. Como en el caso descrito, no es infrecuente observar en el trascurso de los años posteriores la presencia de atrofia distal en los miembros superiores por compromiso de las neuronas motoras medulares de localización cervical. Estos casos pueden plantear diagnósticos diferenciales, como la esclerosis lateral amiotrófica juvenil. De hecho, existen reportes que describen a pacientes que reunían criterios de esclerosis lateral amiotrófica juvenil recesiva (sin los hallazgos típicos de la PEH-ACC) y presentaban mutaciones en SPG1114.
También deberían considerarse subtipos de neuropatía motora distal hereditaria por mutaciones en BSCL2 (seipin)15 en aquellos casos donde existen elementos piramidales asociados y no hay claros antecedentes familiares, indicando mutaciones de novo.
De la misma manera, la ACC puede ponerse de manifiesto años posteriores al inicio de los síntomas motores y, por este motivo, puede ser útil repetir la RM a fin de clarificar el diagnóstico. Rajakulendran et al. consideran que la ACC y del cerebelo podría constituir un marcador fiable de la severidad del cuadro clínico16.
La presencia de cambios en la sustancia blanca periventricular y la atrofia cortical puede variar desde cuadros de afectación incipiente a formas difusas que pueden plantear el diagnóstico diferencial con ciertas leucodistrofias17.
Los hallazgos electrofisiológicos en este caso están en concordancia con descripciones previas de la literatura18. Sin embargo, los mismos hacen difícil establecer topográficamente la causa primaria del fenómeno degenerativo, si se produjeron por un compromiso selectivo primario del soma neuronal de la segunda motoneurona o más bien si estos se dieron por degeneración retrógrada a partir del daño primario de nervio motor.
Como se mencionó previamente, este caso describe 2 nuevas variantes en SPG11 c.7000G>C, p.A2334P (exón 39) c.3146-1G>C (intrón17-18) con segregación familiar (fig. 3 B) y predictivas de ser patogénicas, no descritas previamente en la literatura.
El papel fisiopatogénico de espastacsina y espastizina (SPG11) es aún incierto, aunque se cree que estaría vinculado a mecanismos de transporte axonal. Su expresión es difusa en el sistema nervioso, sobre todo en la corteza cerebral, el hipocampo, el cerebelo y la glándula pineal19. Estudios recientes en el pez cebra han demostrado el papel fisiopatogénico fundamental de esta proteína en el desarrollo normal de la arborización sináptica de las motoneuronas de la médula espinal a nivel de la unión neuromuscular. La parcial depleción proteica de ambas proteínas, spatacsina (SPG11) y spastizina (SPG15), ha demostrado en modelos animales la estrecha relación que se establece entre ambas y explicaría el fenotipo similar en ambos casos20.
En conclusión, este caso describe los hallazgos típicos en la PEHC asociada a ACC debido a mutaciones en SPG11 no reportadas previamente. La presencia de signos de compromiso de motoneurona inferior (atrofia y debilidad distal en los miembros superiores) asociada neuropatía axonal motora pura, ataxia y deterioro cognitivo es un hallazgo habitual. La presencia de ACC en el contexto de un cuadro de PEHC, sea como signo temprano o tardío, indica en primera instancia mutaciones en este gen.
