




Recientes trabajos mostraron la presencia de sintomatología del síndrome autoinflamatorio periódico asociado al receptor tipo 1 para el factor de necrosis tumoral (TRAPS) en pacientes con esclerosis múltiple (EM) portadores de la mutación R92Q en el gen TNFRSF1A. Nuestro objetivo fue investigar la prevalencia de esta mutación en una población argentina de pacientes con EM, describir las manifestaciones clínicas expresadas en estos sujetos y analizar el rol de la misma como factor de susceptibilidad para EM en un estudio de epidemiología molecular.
Pacientes y métodosSe investigó la prevalencia de la mutación R92Q mediante PCR-RFLP en una población de 90 pacientes con EM y 78 controles sanos. Se describieron las características clínicas de ambas patologías (EM y TRAPS) en los portadores de la anomalía genética. Se compararon las frecuencias mutacionales entre casos y controles. Se analizaron variables descriptivas del curso clínico y terapéutico de la EM en el grupo de enfermos estratificado según la presencia de la mutación.
ResultadosCinco pacientes (5,5%) son portadores de la mutación TNFRSF1A R92Q. Presentaron un curso clínico y terapéutico característico de su patología neurológica, habiendo experimentado cuatro de ellos sintomatología sugestiva de TRAPS previo al inicio de la EM. La mutación R92Q fue más frecuente en la población de enfermos que en los controles sanos (1,3%), sugiriendo que esta podría aumentar el riesgo para padecer EM en 4,5 veces aproximadamente.
ConclusionesIdentificamos en nuestra población 5 pacientes con EM y TRAPS portadores de la mutación R92Q en TNFRSF1A. Esta mutación podría ser uno de los distintos factores de susceptibilidad genéticos implicados en el desarrollo de la EM.
Recent studies have recognized a role for TNFRSF1A mutations in Multiple Sclerosis (MS). A number of patients presenting the coexistence of TRAPS (caused by R92Q mutation) and MS were reported. Gene variants in TNFRSF1A might be a genetic risk factor to develop MS. Our aims were: to assess the frequency of TNFRSF1A R92Q mutation in a cohort of MS Argentinean patients and to investigate the role of this mutation in MS clinical characteristics. Secondarily, to investigate the role of this mutation as a genetic risk factor to develop MS.
Patients and methodsWe investigated in a cohort of 90 MS patients from Argentina the TNFRSF1A R92Q mutation by means of a PCR-RFLP assay. TRAPS symptoms, MS clinical characteristics and treatment response and tolerability were investigated in carriers and non-carriers. Secondarily, 78 healthy controls were genotyped to assess the role of this mutation as a risk factor following a case-control study design.
ResultsFive patients (5.5%) carried the R92Q mutation. Four of them reported symptoms suggestive of TRAPS previous to MS onset. No differences in MS clinical features and treatment response and tolerability were found between carriers and non-carriers. R92Q mutation was more frequent in patients than controls increasing the risk to develop MS in about 4.5 times.
ConclusionsThe TNFRSF1A R92Q mutation is not an infrequent finding in MS Argentinean patients that seem to present the coexistence of TRAPS and the demyelinating disease. This genetic variant might be a risk factor to develop MS.
Hacia la mitad del siglo XX se podía leer que “la cuestión de la importancia de los factores hereditarios en la esclerosis múltiple es un tópico controvertido sin poder alcanzar un acuerdo al respecto”1. Sin embargo, los continuos y crecientes avances en el conocimiento de la patología han acabado con esta controversia, afirmándose la existencia de un rol etiopatogénico para distintos factores genéticos2. En este sentido, recientemente Goodin propuso un modelo de la etiopatogenia del trastorno, donde se combina el efecto de los factores genéticos junto al de determinados factores ambientales3. La susceptibilidad genética es el factor determinante más importante de la fisiopatogenia de la esclerosis múltiple.
El síndrome autoinflamatorio periódico asociado al receptor tipo 1 para el factor de necrosis tumoral (TRAPS, por sus iniciales en inglés) es un trastorno hereditario monogénico causado por mutaciones en el gen codificante del receptor de 55 kd para TNFα (TNFRSF1A)4. Característicamente, este trastorno se presenta con episodios recurrentes y autolimitados de fiebre, dolor abdominal, mialgias, erupciones cutáneas, artralgias, faringitis y conjuntivitis5. De las más de 50 mutaciones diferentes identificadas a la fecha, hay una que es la que se encuentra con más frecuencia: la sustitución de una guanina por una citosina que resulta en el cambio de una arginina por una glutamina en la posición aminoacídica 92 (R92Q)6.
La identificación de unos pocos pacientes que, con diagnóstico de esclerosis múltiple (EM) y mutaciones en TNFRSF1A expresaban algunos de los síntomas característicos de TRAPS7–9, permitió plantear las hipótesis alternativas de la coexistencia de dos patologías autoinflamatorias (EM y TRAPS) o la de una única entidad nosológica con manifestaciones sistémicas y compromiso del sistema nervioso central (TRAPS con manifestaciones tipo EM)10. La existencia de agentes terapéuticos con acción específica en la vía de señalización del TNFα11 torna relevante esta controversia en un terreno que podría exceder el meramente fisiopatológico. En consecuencia, con el objetivo de avanzar en el conocimiento del rol de las alteraciones en el gen TNFRS1A en la EM, investigamos la prevalencia de la mutación R92Q en una población argentina de pacientes con EM, describimos las manifestaciones clínicas expresadas por aquellos sujetos que presentan esta mutación y analizamos el rol de la misma como factor de susceptibilidad para el desarrollo de EM en un estudio de epidemiología molecular.
Pacientes y métodosSujetos participantesEl presente estudio incluyó 90 pacientes con diagnóstico de EM recaída remisión consecutivamente atendidos en el Consultorio de Neuroinmunología del Hospital J.M. Ramos Mejía de Buenos Aires, Argentina. La población analizada forma parte de un estudio de investigación de los factores genéticos en la EM iniciado en junio de 2005. Un consentimiento informado, aprobado previamente por la Comisión de bioética del centro hospitalario, fue tomado a cada uno de ellos antes de su inclusión. El diagnóstico de EM fue hecho en función de los criterios de McDonald12, a partir de las características clínicas, sustentado por los hallazgos de las imágenes por resonancia magnética (IRM) y/u otros estudios complementarios. En el momento de la incorporación al estudio se recabaron las siguientes características: edad y sexo; edad y características de la primera recaída de la enfermedad; número de recaídas experimentadas; grado de discapacidad mediante escala EDSS; historia farmacológica con evaluación de la respuesta terapéutica y la tolerabilidad e historia familiar para EM. Posteriormente se interrogó sobre la presencia de síntomas característicos de TRAPS a aquellos sujetos que fueron heterocigotos para la mutación R92Q en el gen TNFRSF1A, e indirectamente sobre la presencia de los mismos en sus familiares de primer grado.
Por otro lado se incluyó una población de 78 controles voluntarios que carecían de antecedentes de patologías del sistema nervioso central o reumatológicas. Estos forman parte de la misma área geográfica que los pacientes y son comparables en términos de origen étnico, edad y sexo. Las muestras provenientes de estos pacientes sirvieron para establecer una comparación entre la prevalencia mutacional en la población de enfermos con respecto a la de sanos, siguiendo un diseño clásico de los estudios de epidemiología molecular caso-control.
Genotipificación de TNFSF1AA todos los sujetos participantes se les extrajo 10ml de sangre venosa mediante venopunción. Se purificó ADN genómico total mediante la utilización del kit FLEXIGENE, siguiendo las instrucciones del fabricante (Qiagen, Hilden, Germany).
Este ADN se diluyó en una solución compuesta por 20mM Tris–HCl (pH 8,8), 50mM KCL, 1,5mM Cl2Mg, dNTPs a 0,2mM cada uno, 50 pM de cada prímer y 1,2 U de Taq polimerasa en un volumen final de 40μl. Mediante reacción en cadena de la polimerasa (PCR) se amplificó un fragmento de 428 pares de bases flanqueante al exón 4 de TNFRSF1A bajo las siguientes condiciones de amplificación: 30s a 95° C, 45s a 60° C y 45s a 72° C por 35 ciclos. Se utilizó el siguiente par de prímers:
5-GGGACACTGCATGGATGTGAG y 5-ACAGAGGAAGTGACGAGGGACA.
Posteriormente, el producto de la amplificación fue sometido a una reacción de restricción utilizando la enzima MspI (New England Biolabs). Normalmente (genotipos no mutados) la digestión del producto de amplificación mediante MspI resulta en fragmentos de 36, 155 y 237 pares de bases. En cambio, cuando se encuentra presente la mutación R92Q se pierde un sitio de reconocimiento para MspI, obteniéndose en consecuencia fragmentos de 191 y 237 pares de bases. Los productos de las digestiones se resolvieron mediante una corrida electroforética en agarosa al 2,5% y visualización mediante tinción con bromuro de etidio bajo luz ultravioleta. De esta forma fue posible identificar la presencia o ausencia de la mutación R92Q en cada una de las muestras analizadas.
Todas las reacciones de genotipificación se realizaron de forma ciega para el origen de las muestras (casos o controles). Aquellas muestras que resultaron positivas para la mutación investigada fueron confirmadas en una segunda reacción.
Análisis estadísticoEl equilibrio de Hardy-Weinberg fue probado con test exacto en la población de controles. Para el análisis de TNFRSF1A como factor de susceptibilidad de EM se definió un modelo de herencia dominante para el alelo R92Q, de acuerdo con la segregación descrita en las familias con TRAPS secundarias a esta mutación4, investigándose la asociación entre la mutación R92Q y el desarrollo de EM mediante la prueba de Chi cuadrado.
Para el análisis del rol de la mutación R92Q en el curso clínico de la EM se compararon las características recabadas (mencionadas en el apartado sujetos participantes) mediante modelos de regresión logística o lineal, según correspondiese, entre el grupo de pacientes portadores de la mutación y los que no lo eran.
Se estableció como nivel de significación p< 0,05. Se utilizó como herramienta informática el paquete estadístico STATA versión 9.
ResultadosCaracterización de los pacientes con esclerosis múltiple portadores de la mutación R92Q en el gen TNFRSF1A- 1.
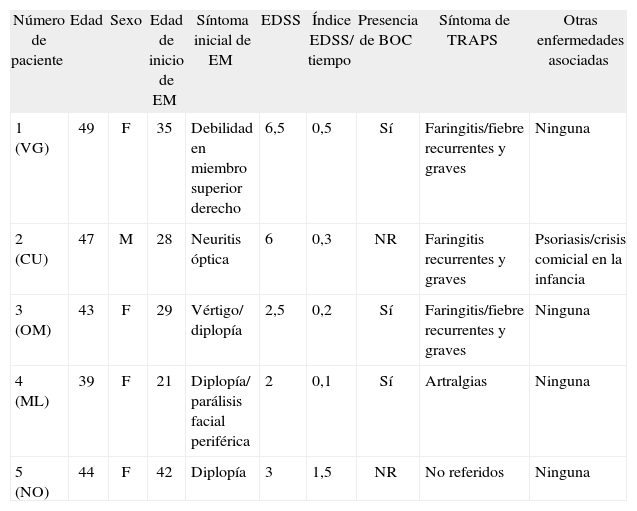
Cuadro clínico de EM: cinco pacientes son portadores de la mutación TNFRSF1A R92Q, indicando una prevalencia mutacional de un 5,5%; cuatro de ellos son mujeres. En el momento de la inclusión en el estudio 4 de ellos se encontraban en estadios no progresivos, mientras que el restante estaba en un estadio secundariamente progresivo. La edad de comienzo de los síntomas se encontró entre los 21 y 42 años (media = 31), observándose un tiempo medio de 15,4 meses entre la primera y la segunda recaída clínica. El primer evento fue más frecuentemente del tronco encefálico: parálisis facial periférica y diplopía (ML), diplopía y vértigo (OM) y diplopía (NO). Los otros dos pacientes tuvieron como síntoma inicial neuritis óptica (CU) en un caso y debilidad de un hemicuerpo en el otro (VG). Una de las pacientes (OM) presentó parálisis facial periférica aislada 12 años antes del comienzo de la enfermedad (tabla 1). La duración promedio de enfermedad hasta el momento de la inclusión fue de 13,4 años (rango: 2–19).
Tabla 1.-Características de los pacientes portadores de la mutación R92Q
Número de paciente Edad Sexo Edad de inicio de EM Síntoma inicial de EM EDSS Índice EDSS/ tiempo Presencia de BOC Síntoma de TRAPS Otras enfermedades asociadas 1 (VG) 49 F 35 Debilidad en miembro superior derecho 6,5 0,5 Sí Faringitis/fiebre recurrentes y graves Ninguna 2 (CU) 47 M 28 Neuritis óptica 6 0,3 NR Faringitis recurrentes y graves Psoriasis/crisis comicial en la infancia 3 (OM) 43 F 29 Vértigo/ diplopía 2,5 0,2 Sí Faringitis/fiebre recurrentes y graves Ninguna 4 (ML) 39 F 21 Diplopía/ parálisis facial periférica 2 0,1 Sí Artralgias Ninguna 5 (NO) 44 F 42 Diplopía 3 1,5 NR No referidos Ninguna BOC: bandas oligoclonales en líquido cefalorraquídeo; EDSS: Expanded Disability Status Scale; EM: esclerosis múltiple; NR: no realizado.
- 2.
Gravedad clínica: el número de recaídas total fue de 44 para los 67 años totales de historia de enfermedad en los 5 pacientes. Sólo uno tuvo un promedio de recaídas mayor a una por año con un índice EDSS/tiempo de 1,5. El resto tuvieron índice EDSS/tiempo de 0,5 o menor. En consecuencia, estos parámetros no parecen sugerir un curso más agresivo que el habitualmente observado en la patología.
- 3.
Tratamiento de la EM: todos recibieron tratamientos modificadores de la enfermedad, inmunomoduladores y/o inmunosupresores. En el momento del estudio 3 recibían IFN β 1a intramuscular, uno IFN β 1a subcutáneo y el último acetato de glatiramer. Todos tuvieron buena respuesta al tratamiento instaurado y en ningún caso se debió modificar el mismo por falta de respuesta o efectos adversos serios. Sólo en un paciente (CU) se rotó IFN β1a subcutáneo por IFN β 1a intramuscular debido a eritema y dolor en el sitio de punción. Dos pacientes con IFN refirieron la presencia de manifestaciones tipo pseudogripales al inicio del tratamiento. No presentaron otros efectos adversos a la medicación.
- 4.
Síntomas de TRAPS: los pacientes fueron interrogados sobre síntomas de TRAPS de forma directa, excepto uno, cuyos datos se extrajeron de la historia clínica. Los síntomas de TRAPS recabados consistieron en faringitis de repetición en tres pacientes (con amigdalectomía también en los tres), fiebre recurrente en la infancia en dos, artralgias en uno y fiebre desencadenada por herpes bucal en otro. Ninguno tuvo signos o síntomas mayores de TRAPS tales como artritis, sinovitis o fallo renal. En todos los casos los síntomas de TRAPS aparecieron antes del inicio de la EM. No hubo presencia de colagenopatía asociada ni de otra enfermedad autoinmune, con la excepción de psoriasis en un caso.
- 5.
Familiares: se interrogó sobre los familiares de primer grado; estos tampoco presentaron signos o síntomas mayores de TRAPS. Una familia refirió faringitis y conjuntivitis de repetición (presentes en 4 de los 6 familiares de primer grado del caso índice). Resulta destacable que el padre de la paciente que había presentado parálisis facial periférica varios años antes del inicio de la EM tuvo también parálisis facial periférica recurrente (tres episodios).
Observamos una mayor probabilidad de presentar, como primera recaída de la enfermedad, un evento con sintomatología correspondiente a la afectación de las estructuras del tronco del encéfalo en los sujetos portadores de la mutación R92Q, que en los no portadores (p = 0,03; OR = 7,5). Por el contrario, no encontramos diferencias en ninguna de las otras características de la patología analizadas entre aquellos sujetos portadores de la mutación R92Q y los no portadores; es decir, no hubo diferencias significativas en el número de recaídas experimentadas, en el periodo transcurrido entre la primera y la segunda recaída, en las variables de gravedad de la enfermedad o en el grado de discapacidad de la misma.
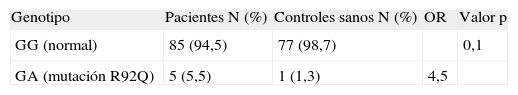
Análisis del rol de la mutación R92Q como factor de susceptibilidad para el desarrollo de esclerosis múltipleCon el objetivo de investigar la hipótesis de un rol de la mutación R92Q en el riesgo de desarrollar EM, realizamos un estudio de epidemiología molecular con un diseño de casos y controles comparando las prevalencias de la mutación en una población de enfermos con la de una de controles sanos. La distribución de genotipos obtenida en las 168 muestras analizadas (90 casos y 78 controles) se resume en la tabla 2. Se puede observar una tendencia para una frecuencia mutacional mayor en la población de sujetos con EM que en los controles sanos (p = 0,1). Otra manera de expresar lo propio es en términos de riesgo, sugiriendo nuestros resultados que ser portador de la mutación R92Q aumenta el riesgo para padecer EM en 4,5 veces aproximadamente. La distribución de genotipos en los controles se encontró en equilibrio de Hardy-Weinberg (p = 0,95).
DiscusiónEl presente trabajo permitió identificar 5 pacientes con EM portadores de la mutación más frecuentemente causante del síndrome autoinflamatorio hereditario TRAPS4 que presentan, junto a un curso clínico característico de su patología de base, el antecedente de sintomatología recurrente sugestiva de un desorden inflamatorio sistémico familiar. Estos pacientes son los primeros identificados en nuestra población y se suman a sólo otros 21 casos reportados en la literatura7,9. Estos últimos provienen de una serie investigada en una población alemana. Además, resulta destacable la observación en nuestros pacientes de una alta frecuencia de afectación de las estructuras del tronco encefálico al inicio de la enfermedad, y en particular el compromiso del nervio facial en dos casos, con recurrencia familiar de esta afectación en uno de ellos. El compromiso periférico del nervio facial al inicio de la EM es muy poco frecuente. Dos pequeños trabajos lo encontraron en aproximadamente un 5% de los pacientes13,14. En cambio, además de los dos pacientes de nuestra casuística, otros tantos presentaron este compromiso en la serie de 21 pacientes alemanes7. En consecuencia, la frecuencia del compromiso inicial del nervio facial en los pacientes con EM y la mutación R92Q sería aproximadamente tres veces mayor (15,4%) que en la población general de pacientes con EM. Sin embargo, el número pequeño de pacientes descritos impide considerar esta observación más que como generadora de hipótesis. En este sentido, un rol para el TNFα en la parálisis facial periférica idiopática fue sugerido por algunos autores que encontraron niveles séricos aumentados de esta citocina en pacientes con dicha patología15. Por el contrario, otros autores fallaron al encontrar similares hallazgos16, y por lo tanto no está claro el rol del TNF en esta neuropatía.
Por otro lado, los resultados de nuestro estudio de epidemiología molecular pueden sugerir en nuestra población un rol para la mutación R92Q en el gen TNFRSF1A como factor de riesgo genético para el desarrollo de EM. Aunque este hallazgo es producto del análisis de una población relativamente pequeña de pacientes, que en consecuencia brinda baja potencia para alcanzar significación estadística a las diferencias observadas, sí que es concordante con los resultados de un estudio similar recientemente realizado en una población de más de 2.000 pacientes, y con los del metaanálisis de estudios de asociación a genoma completo que incluyeron a más de 9.000 sujetos17. En consecuencia, creemos que la mutación R92Q en TNFRSF1A podría ser considerada un factor de susceptibilidad para el desarrollo de EM. Cabe mencionar que otros autores, pese a encontrar una mayor prevalencia de la mutación R92Q en los pacientes con EM que en la población general, también fallaron en demostrar significación estadística a consecuencia del limitado número de pacientes analizados en sus respectivas investigaciones18. Esta situación es frecuentemente observada en el campo de la genética compleja de las enfermedades prevalentes, donde reiteradamente se le atribuye un rol causal en los resultados discordantes de esta clase de estudios19.
Tradicionalmente el acento en la fisiopatogenia de la EM estuvo puesto en la desregulación de los mecanismos de la respuesta inmune T o adaptativa20,21. Sin embargo, en los últimos años se ha mostrado una creciente acumulación de evidencia que apoya el importante rol de las alteraciones en la inmunidad innata en la compleja patogenia del trastorno. En este sentido pueden mencionarse: a) las alteraciones observadas en la regulación de células dendríticas y el propuesto posible mecanismo de acción del interferón beta a este nivel22,23; b) la desregulación de las distintas vías de señalización dependientes del interferón endógeno, que sugieren la existencia de un sistema inmune de defensa a virus preactivado constitutivamente (estado hiperinflamatorio) en un subgrupo de pacientes con EM24 y c) la mayoría de los factores de susceptibilidad genéticos para el desarrollo de la EM recientemente identificados se encuentran en genes codificadores de moléculas involucradas en la inmunidad innata17. El TNFα es una citocina pleotrópica con distintos roles en la regulación de la respuesta inmune innata25. Distintas observaciones sustentan un rol de esta citocina en la fisiopatogenia de la enfermedad: a) niveles elevados han sido encontrados en sueros de pacientes con EM en actividad26; b) el tratamiento con anticuerpos monoclonales anti-TNFα ha resultado en la exacerbación de lesiones desmielinizantes en algunos pacientes27,28 y c) variantes en la secuencia del gen codificante de TNFα fueron indicadas como factores de riesgo para el desarrollo de EM en algunas poblaciones29. Además, el síndrome TRAPS podría también representar un estado hiperinflamatorio similar a lo sugerido más arriba para la fisiopatología de la EM. Este estado hiperinflamatorio podría ser consecuencia de la desregulación en los niveles circulantes de TNFα, que resultan de fallos en la secreción de su receptor soluble (codificado por TNFRSF1A) motivados por la mutación R92Q6. Resulta, entonces, plausible considerar la señalización mediada por TNFα como uno de los mecanismos implicados en ambos trastornos y, en consecuencia, representa una hipótesis fisiopatogénica de los resultados clínico-epidemiológicos de nuestra investigación que deberá evaluarse en futuros estudios experimentales.
En conclusión, hemos identificado en nuestra población de pacientes con EM un número de sujetos que presentan una mutación en el gen codificante del receptor soluble para TNFα y sintomatología característica del síndrome TRAPS que es causado por esta alteración genética. Aunque ambos trastornos parecen comportarse como clínicamente independientes, sí podrían compartir mecanismos fisiopatogénicos comunes. La mutación analizada además podría ser uno de los distintos factores de susceptibilidad genéticos implicados en el desarrollo de la EM. Estas observaciones ilustran el rol del TNFα en la fisiopatogenia de esta enfermedad desmielinizante.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
