




En el presente documento se hace el reporte de un caso de síndrome de Guillain y Barré variedad Miller Fisher asociado a hepatitis B aguda, el cual no mejoró pese al tratamiento con inmunoglobulina, lo que se asoció al cuadro hepatítico agudo y elevada carga viral. Tras recibir el tratamiento antiviral, tanto el cuadro hepático como el neurológico remitieron totalmente sin secuelas.
In this paper we report a case of Guillain Barré syndrome Miller Fisher variety associated with acute hepatitis B, which did not improve despite treatment with immunoglobulin, which was associated with the acute hepatitic picture and high viral load. After receiving antiviral treatment, both the liver and neurological symptoms completely disappeared without sequelae.
El síndrome de Guillain y Barré (SGB) constituye un conjunto de entidades clínicas que se manifiestan con diferentes subtipos clínicos con rasgos electrofisiológicos y anatomopatológicos distintos, cuyo máximo pico clínico de afectación se sitúa entre la 2.a y la 4.a semanas de evolución desde el inicio de los síntomas; se caracteriza clínicamente por la presencia de una parálisis flácida aguda con arreflexia, trastorno sensorial variable y elevación de las proteínas en el líquido cefalorraquídeo. Es considerado como la causa más frecuente de parálisis flácida, con una incidencia mundial reportada de 0,6-4 por 100.000 habitantes por año. Suele afectar a personas de cualquier edad y sexo con 2 picos de presentación: uno en la etapa adulta joven (15-34 años) y otra en ancianos (60-74 años); es rara su presentación en niños menores de un año de edad1.
En pacientes con SBG, en quienes se identificó el antecedente de un proceso infeccioso, se asociaron los siguientes microorganismos:
- •
Campylobacter jejuni: 20-50%.
- •
Citomegalovirus: 5-22%.
- •
Haemophylus influenzae: 2-13%.
- •
Epstein-Barr: 10%.
- •
Mycoplasma pneumoniae: 5%.
En menor proporción se ha reportado: borreliosis de Lyme, hepatitis de tipos A, B, C y E, fiebre tifoidea, dengue, influenza A, virus de Zika, chikungunya y VIH. Otras condiciones asociadas: cirugías, vacunas (implicadas las de hepatitis B) y traumatismos1.
Antes del advenimiento de los arbovirus chikungunya y Zika en la región de las Américas, el virus del dengue condicionaba cuadros neurológicos agudos como Guillain y Barré en forma esporádica. Sin embargo, durante las oleadas de 2014 y 2015 hubo una incidencia incrementada de afectación neurológica aguda, en menor proporción por chikungunya y con la mayor incidencia asociada al virus de Zika. Actualmente se considera al virus de Zika un virus neurotrópico que ha condicionado casos de síndrome congénito por Zika (del cual destaca la microcefalia) y SBG2,3.
El síndrome de Miller Fisher es la variante atípica más frecuente del SGB. Se caracteriza por la tríada clásica de oftalmoplejía, ataxia y arreflexia. El componente motor o sensorial tiene menos relevancia, y se declara en el 5% de los casos. Algunas veces puede manifestarse con parálisis bulbar o facial y ser precedido de la infección por Campylobacter jejuni. El curso de este síndrome es bueno, con recuperación temprana y completa. En el síndrome de Miller Fisher se desarrolla mimetismo molecular, el cual condiciona la aparición de anticuerpos contra gangliósidos, predominantemente los gangliósidos GM1 que predominan en el nervio óptico y en los oculomotores, casi siempre secundarios a un proceso infeccioso enteral. La subclase de inmunoglobulina G, que se produce y es la causante de las manifestaciones propias de este síndrome, es un anti-GQ1b (inmunoglobulina G1), aunque también se pueden expresar otros como GM1b, GD1a y GalNAc-GD1a.
Se recomienda cumplir los criterios de Brighton para un diagnóstico más preciso del síndrome1,4.
Respecto del tratamiento, los pilares siguen siendo la inmunoglobulina intravenosa y la plasmaféresis. Pocos pacientes llegan a requerir asistencia mecánica ventilatoria y estancia en unidad de cuidados intensivos, aunque en cada caso el riesgo de necesitar manejo invasivo es latente1.
Presentamos el caso de un paciente masculino joven afectado por SBG en su variedad Miller Fisher, durante la oleada de virus de Zika del 2016 en el que, sin embargo, no se demostró asociación etiológica con esta entidad, por lo que se amplió el estudio infectológico del caso, y se identificó hepatitis B activa. Pese al tratamiento inicial con inmunoglobulina intravenosa, no hubo mayor impacto en el estado clínico y pronóstico del paciente, quien presentó una elevada carga viral, que era la condicionante del cuadro, que se resolvió tras inicio de entecavir y llevar a la negatividad la carga viral.
Caso clínicoPaciente masculino de 32 años de edad, quien presentó SBG variedad Miller Fisher, con la tríada de oftalmoplejía bilateral, ataxia y arreflexia, 3 meses antes de su ingreso (septiembre de 2016). Se le realizó punción lumbar, que demostró disociación albuminocitológica, así como TAC craneal, reportada como normal. También se le realizó estudio electrofisiológico compatible con polirradiculopatía axonal difusa (se catalogó como Brighton 1, cumpliendo los criterios para diagnóstico). Recibió inmunoglobulina intravenosa (IgIV) por 5 días a razón de 0,4g/kg/día. El paciente no presentó mejoría, persistiendo en clase 4 de Hugues desde su inicio hasta los 3 meses posteriores, pese a inmunoglobulina y a terapia física y rehabilitación.
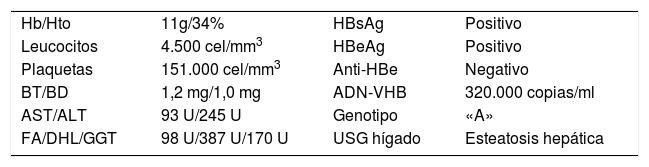
Se le realizó determinación de PCR-RT para dengue, chikungunya y virus de Zika, que fueron negativas. Asimismo, el análisis coprológico y el coprocultivo fueron negativos para Campilobacter jejuni y otras enterobacterias. Se extendió el estudio infectológico del caso con ELISA para VIH, que fue negativo, y se solicitó panel viral de hepatitis B y C, que resultó positivo para hepatitis B (HBeAg+ y HBsAg+). Se complementó el estudio con carga viral, la cual se reportó en 320.000 copias/ml.
Con la evidencia anterior, se inició tratamiento médico a base de entecavir (1mg/día), el cual se mantuvo por 6 meses, con posterior envío a gastroenterología para seguimiento. Presentó buena evolución, con carga viral indetectable a los 2 meses de tratamiento y negatividad del antígeno e. Asimismo, la clase de Hugues que se alcanzó fue 1, no ameritó ningún nuevo tratamiento para Guillain Barré y actualmente el paciente ha retornado a sus actividades habituales, sin secuela neurológica ni oftálmica (tabla 1).
DiscusiónLa etiología del SBG a menudo no se precisa y se enfoca su abordaje más al diagnóstico y tratamiento. Durante la oleada de virus de Zika del 2016, se identificaron incidencias incrementadas de este síndrome gracias al neurotropismo viral del Zika, principalmente. Sin embargo, en la mayoría de los casos no fue posible demostrar la relación causal directa del SGB con Zika: existen otros virus que de igual forma tienen neurotropismo, tal es el caso de los virus de las hepatitis B y C, de los que existen reportes aislados de enfermedad neurológica, incluido el SGB. En el presente caso destacó que, pese al tratamiento específico con IgIV, no hubo mejoría clínica ni remisión de la enfermedad, la cual estaba supeditada a la carga viral excesivamente elevada. Se demostró mejoría del síndrome tras el tratamiento específico de la causa desencadenante (entecavir es uno de los principales tratamientos orales de primera línea para hepatitis B), lo cual hace ver la importancia de identificar el agente de cada caso de SGB para un tratamiento específico y oportuno que, aunado a la IgIV o a la plasmaféresis, puedan mejorar la oportunidad de una remisión completa y la curación de la enfermedad5-8.
Respecto de la hepatitis viral como causa de síndromes neurológicos agudos, incluido el SGB, existen reportes de casos aislados que involucran a hepatitis de los tipos A, B, C, D y E. Respecto de la hepatitis B, hasta 2013 se habían identificado en la bibliografía poco más de 20 casos documentados del SGB asociados a hepatitis B activa y existe reporte de recaídas del SGB asociadas a reactivación o exacerbación de la hepatitis B crónica. De igual forma, hay múltiples casos reportados asociados a vacunación contra hepatitis B5.
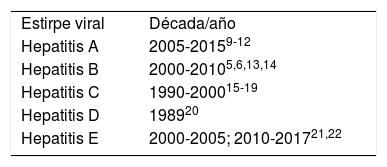
Las otras estirpes de hepatitis virales han tenido implicación como causales de SBG: destacamos que cada tipo ha tenido periodos de mayor incidencia y que algunos no se han vuelto a identificar, tal es el caso de la hepatitis delta o D, la cual actualmente se reporta como una coinfección asociada a hepatitis B. La hepatitis C tuvo su mayor auge durante la década de los noventa, temporada en la que se reportaron la mayoría de los casos asociados a dicha estirpe viral. Las hepatitis A y B tuvieron la mayor incidencia de casos asociados a SGB entre el año 2000 y el 2015. Actualmente se está viendo una mayor incidencia de casos asociados a hepatitis E, de la cual se menciona que es neurotrópica además de hepatotrópica (tabla 2).
En conclusión, se recomiendan los siguientes aspectos:
- 1.
Sospechar etiología viral hepatítica en casos con afectación hepática aguda o crónica, principalmente en aquellos con elevación de ALT más de 3 veces el valor normal o patrón colestásico biliar, asociado a la parálisis flácida aguda.
- 2.
Tomar en cuenta a las hepatitis virales (A-E) como potenciales agentes causales del SGB, considerando el pico de incidencia actual de hepatitis E.
Los autores declaran no tener ningún conflicto de intereses.
