



We are writing about an unusual case, to our knowledge not previously reported, of sudden neurological deterioration and coma.
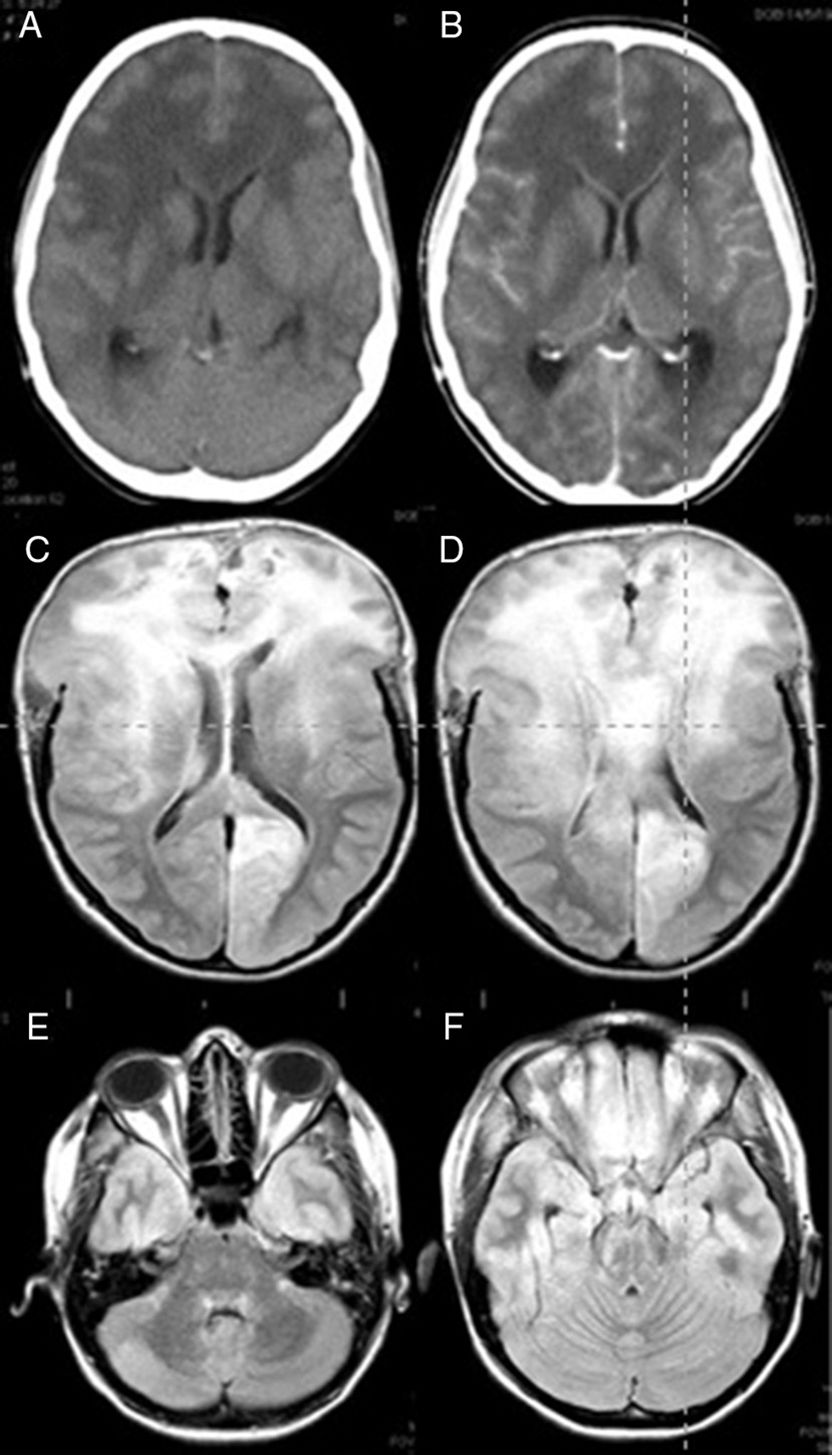
The case is that of a 25-year old woman who had given birth 3 months before after a managed full-term pregnancy without complications. She was treated with oral iron due to postpartum anaemia but had no other relevant medical history. This patient arrived at the ER presenting with a one-month history of headaches, apathy, and bradypsychia. The headaches had worsened in the last 48 hours with incomplete response to conventional analgesic treatment. Nausea and vomiting had appeared in the last few hours. She had no fever. While in the ER she had a sudden deterioration in level of consciousness together with a tonic–clonic seizure. She was intubated. An emergent CT scan showed signs of diffuse cerebral oedema and obliterated subarachnoid cisterns. Leptomeningeal and ependymal enhancement was present (Fig. 1A and B). These findings were suggestive of cerebritis accompanied by meningeal involvement and ventriculitis.
 and FLAIR MRI (C–F) images. (A) Predominant presence of diffuse white matter hypodensity in both frontal lobes and involvement of the corpus callosum indicating vasogenic oedema. Convexity sulci effacement is seen. (B) Image was taken after the administration of contrast, showing no focal enhancement. (C) Signs of intracranial hypertension involving both frontal lobes and the corpus callosum are present. (D) Brain herniation persists despite extensive bilateral frontal craniectomy. (E and F) Infratentorial diffuse involvement is also present.")
Brain CT (A and B) and FLAIR MRI (C–F) images. (A) Predominant presence of diffuse white matter hypodensity in both frontal lobes and involvement of the corpus callosum indicating vasogenic oedema. Convexity sulci effacement is seen. (B) Image was taken after the administration of contrast, showing no focal enhancement. (C) Signs of intracranial hypertension involving both frontal lobes and the corpus callosum are present. (D) Brain herniation persists despite extensive bilateral frontal craniectomy. (E and F) Infratentorial diffuse involvement is also present.
She was admitted to the ICU and 6 hours later suddenly developed non-reactive bilateral mydriasis. A decompressive wide bifrontal craniectomy with bilateral decompression of the frontal and temporal lobes was carried out as a compassionate treatment, and a biopsy of the right frontal lobe was performed. During the intervention, we observed the brain to be congested and of a hard consistency. After the procedure an intracranial pressure sensor was placed which initially recorded pressures below 15mmHg. After intervention mydriasis was reversed and pupillary reflexes were restored. CSF analysis showed no cytochemical abnormalities and the results from the CSF culture were negative for bacteria, viruses, and fungi. The results of blood serology tests for autoimmune diseases were also negative. A brain MRI showed supra and infratentorial diffuse involvement, especially in the frontal lobes and corpus callosum. Small areas of contrast enhancement and signs of intracranial hypertension were also observed (Fig. 1C–F).
Clinical and radiological differential diagnosis included infectious entities such as progressive multifocal leukoencephalopathy and other forms of viral encephalitis; autoimmune diseases such as acute disseminated encephalomyelitis, vasculitis or connective tissue diseases; metabolic disorders such as certain forms of leukodystrophy; encephalopathy after radiotherapy; and such tumours as gliomatosis cerebri or primary brain lymphoma.1,2
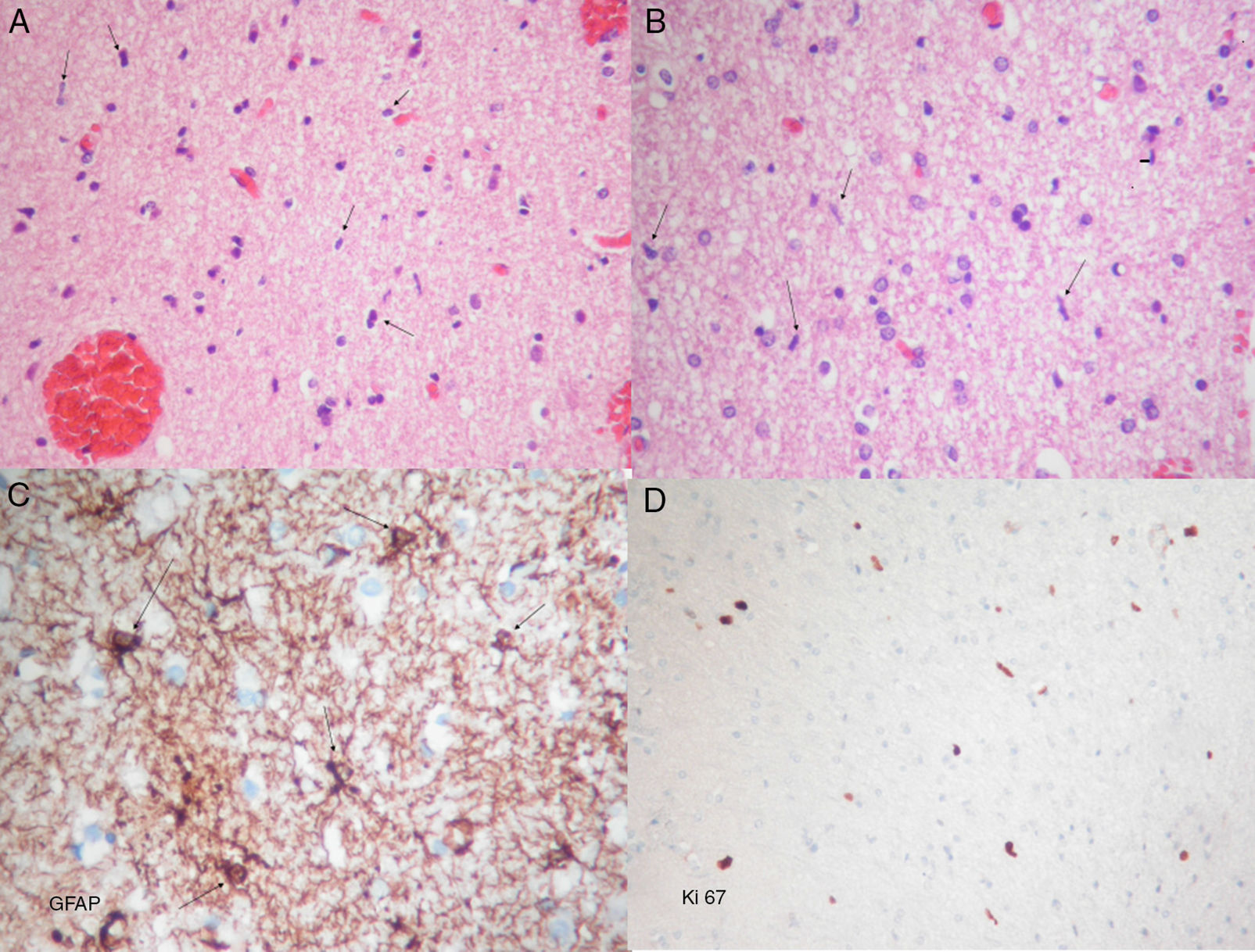
Histopathological examination revealed diffuse astrocyte proliferation and low cell density with features of a low-grade glial tumour (Fig. 2). These findings added to those from the MRI were consistent with gliomatosis cerebri.
 Haematoxylin–eosin stain. Proliferation of moderate cellularity characterised by the presence of naked nuclei (arrows) can be observed. (B) Haematoxylin–eosin stain. Oligodendroglial lineage cells are abundant. Glial elements composed of bare spindle-shaped moderately atypical nuclei can be seen (arrows). (C) Immunohistochemical GFAP (glial fibrillary acidic protein) staining showing atypical astrocytic elements with short irregular coarse extensions (arrows). (D) Ki67 staining showing proliferative activity around 2%. A, B, and D: original magnification 10×; C: original magnification 40×.")
Photomicrographs showing frozen and permanent sections. (A) Haematoxylin–eosin stain. Proliferation of moderate cellularity characterised by the presence of naked nuclei (arrows) can be observed. (B) Haematoxylin–eosin stain. Oligodendroglial lineage cells are abundant. Glial elements composed of bare spindle-shaped moderately atypical nuclei can be seen (arrows). (C) Immunohistochemical GFAP (glial fibrillary acidic protein) staining showing atypical astrocytic elements with short irregular coarse extensions (arrows). (D) Ki67 staining showing proliferative activity around 2%. A, B, and D: original magnification 10×; C: original magnification 40×.
Forty eight hours after surgery the patient experienced an increase in intracranial pressure refractory to treatment. After obtaining family consent, a decision was made to limit treatment. The patient died 6 days after surgery.
Gliomatosis cerebri was first described by Nevin in 19381 and represents just under 1% of all astrocytomas.3,4 It is a neoplastic disorder originating from glial cells,3,5,6 defined by the infiltration of at least 2 lobes.1,3,6 Despite this fact, gliomatosis cerebri typically preserves the macroscopic structure and cytoarchitecture of the CNS.3,6
Although this condition has been reported in children,7 it usually occurs in patients aged between 40 and 50.6 Its incidence appears to be slightly higher in men.3 It is mainly located in the supratentorial level, but it often spreads to infratentorial structures.8–10 The corpus callosum, thalamus, and basal ganglia are frequently involved.6 In addition, expansion to the entire neuraxis has been reported.8
This disease is often overlooked in the early stages, so diagnosis commonly occurs in the advanced stages.7 Common initial symptoms include the appearance of focal neurological deficits or the presence of less specific signs such as headache, nausea, vomiting, seizures, personality changes, or cognitive impairment.5
Neuroimaging findings of gliomatosis cerebri are characteristic but rarely specific.9,10 This implies the need for a broad differential diagnosis. T2 and FLAIR MRI sequences usually reveal hyperintense areas with asymmetric and/or heterogeneous distribution.9,10 The corpus callosum is usually involved and appears thickened.9,10 Loss of differentiation between grey and white matter is also characteristic.9,10 In type I gliomatosis cerebri there is usually no contrast enhancement, while type II commonly displays areas with contrast enhancement that correlate with anaplastic transformation.9,10 Spectroscopy and sequences of relative cerebral blood volume can help in recognising the glial origin of this tumour.3 These sequences can also be useful to more accurately determine areas for biopsy.3
Histopathological features often correspond to low-grade glial tumours,5,6 although tumour progression showing high grade features can take place.5,6 The most common cellular phenotype is astrocytic, but it may also present oligoastrocytic or oligodendrocytic phenotype.5 Despite its histological appearance the clinical behaviour of this tumour correlates to at least grade III in the WHO classification.6
A definitive diagnosis is made with neuroimaging and histopathological features.3,6 Nevertheless heterogeneity in histological findings may hamper the diagnosis.3
Because glioma are highly infiltrative and diffuse, the role of surgery is limited to biopsy for diagnostic purposes.3,4 The mainstays of treatment are radiotherapy11,12 and chemotherapy.13,14 Even so, median survival time is estimated at around 14.5–18 months.14,15 Prognostic factors are similar to those of other gliomas.3
A dismal clinical presentation of gliomatosis cerebri is featured in the current case. To our knowledge, intracranial hypertension syndrome with coma refractory to treatment resulting in fatality had not previously been reported as a presentation of gliomatosis cerebri.
Conflicts of interestThe authors report no conflicts of interest concerning the materials or methods used in this study or the findings described in this paper.
Please cite this article as: Vargas López AJ, Garbizu Vidorreta JM, Salinero Paniagua E, Fernández Carballal C. Una causa excepcional de deterioro neurológico repentino y coma. Neurología. 2018;33:196–199.
