



La enfermedad de Huntington (EH) es un trastorno neurodegenerativo y hereditario. Gracias al diagnóstico predictivo se han descrito características clínicas incipientes en la fase prodrómica.
ObjetivoComparar la ejecución en tareas cognitivas de portadores (PEH) del gen de la huntingtina y no portadores (NPEH) y observar la variabilidad en la ejecución, dependiendo de la carga de la enfermedad y cercanía a la etapa manifiesta (edad de inicio de los síntomas).
MétodoLos 146 participantes de un Programa de Diagnóstico Predictivo de EH (PDP-EH) fueron divididos en PEH (41,1%) y NPEH (58,9%). Mediante fórmulas matemáticas se obtuvo la carga de enfermedad y cercanía a la etapa manifiesta en el grupo PEH y se correlacionó con la ejecución neuropsicológica.
ResultadosSe observaron diferencias significativas entre los grupos con las pruebas Mini-Mental State Examination (MMSE), Stroop-B, SDMT y fluidez fonológica. En el grupo PEH se observaron correlaciones entre la carga de enfermedad con la MMSE, Stroop-B y SDMT. El grupo «Cerca» de la etapa manifiesta es el que obtuvo la puntuación más baja en las pruebas MMSE, Stroop-B, Stroop-C, SDMT y fluidez verbal semántica. De acuerdo al MANCOVA, el efecto MMSE evidencia diferencias estadísticamente significativas entre carga de la enfermedad y la cercanía de inicio de los síntomas.
ConclusionesSe observa un nivel menor de desempeño en el grupo PEH con probabilidad de inicio cercano de la fase manifiesta en pruebas que evalúan la velocidad de procesamiento y atención. La disfunción cognitiva prefrontal se altera de manera precoz varios años antes del diagnóstico motor de la EH.
Huntington disease (HD) is a hereditary neurodegenerative disorder. Thanks to predictive diagnosis, incipient clinical characteristics have been described in the prodromal phase.
ObjectiveTo compare performance in cognitive tasks of carriers (HDC) and non-carriers (non-HDC) of the huntingtin gene and to analyse the variability in performance as a function of disease burden and proximity to the manifest stage (age of symptom onset).
MethodA sample of 146 participants in a predictive diagnosis of HD programme were divided into the HDC (41.1%) and non-HDC groups (58.9%). Mathematical formulae were used to calculate disease burden and proximity to the manifest stage in the HDC group; these parameters were correlated with neuropsychological performance.
ResultsSignificant differences were observed between groups in performance on the Mini-Mental State Examination (MMSE), Stroop-B, Symbol-Digit Modalities Test (SDMT), and phonological fluency. In the HDC group, correlations were observed between disease burden and performance on the MMSE, Stroop-B, and SDMT. The group of patients close to the manifest stage scored lowest on the MMSE, Stroop-B, Stroop-C, SDMT, and semantic verbal fluency. According to the multivariate analysis of covariance, the MMSE effect shows statistically significant differences in disease burden and proximity to onset of symptoms.
ConclusionsMembers of the HDC group close to the manifest phase performed more poorly on tests assessing information processing speed and attention. Prefrontal cognitive dysfunction appears early, several years before the motor diagnosis of HD.
La enfermedad de Huntington (EH) es un trastorno neurodegenerativo y hereditario de transmisión autosómica dominante. Se caracteriza por alteraciones motoras, psiquiátricas y cognitivas. Tiene una prevalencia de 10 pacientes por cada 100.000 personas en países occidentales1. Es causada por la expansión de un trinucleótido repetido CAG en el gen huntingtina (HTT) localizado en 4p16.32,3; se consideran 4rangos de repetidos:<27 repeticiones es el fenotipo normal; de 27 a 35 son alelos intermedios; de 36 a 39 es penetrancia incompleta;>40 supone una penetrancia completa. Estos rangos explican los cambios en las medidas de resultado en sujetos presintomáticos4.
Generalmente, los síntomas clínicos en la EH se manifiestan en la cuarta década de la vida, con un tiempo de evolución de 15 a 20 años3,5. En la actualidad, no existe ningún tratamiento curativo, ni intervención terapéutica que modifique la progresión de la enfermedad6. La evolución de la EH puede estar dividida en 2periodos: presintomático y sintomático. El periodo presintomático consta de 2fases: la presintomática, en la cual los portadores no se distinguen clínicamente de los sanos, y la prodrómica, que se caracteriza por leve sintomatología psiquiátrica, motora y cognitiva7. El periodo sintomático manifiesto empieza formalmente cuando se realiza el diagnóstico clínico motor de la enfermedad; es decir, cuando el paciente presenta alteraciones motoras, que pueden incluir algunos movimientos anormales como corea, bradicinesia, distonía e incoordinación, con una probabilidad ≥99% de que la causa sea consecuencia de la EH8,9.
Según Reilmann et al.7, la fase prodrómica es de particular interés en la investigación clínica debido a que es el momento idóneo para probar tratamientos que retrasen el inicio y modifiquen la progresión de la enfermedad y que tengan el menor número de efectos negativos. Gracias a las pruebas de diagnóstico predictivo para la EH, se han descrito características clínicas incipientes que se presentan en la fase prodrómica y que son posibles marcadores clínicos de la enfermedad10. Algunos de estos marcadores están relacionados con funciones cognitivas. Al comparar ejecuciones en tareas cognitivas de personas en riesgo de desarrollar la EH, se ha observado que los portadores de la mutación tienen un funcionamiento cognitivo menor que los no portadores, principalmente en tareas de velocidad de procesamiento, funciones ejecutivas, atención, memoria episódica y habilidades visoespaciales9,11,12.
Además, existe una correlación inversa entre el número de repetidos y la edad de inicio de la enfermedad: a mayor número de repetidos, menor edad de inicio de los síntomas; esto es, mayor probabilidad de que el deterioro del tejido cerebral, los síntomas motores, cognitivos y la pérdida de la capacidad funcional en la vida diaria se presenten a edad más temprana13.
Se ha tratado de relacionar la progresión de los síntomas cognitivos en portadores de la EH y la carga genética de la enfermedad a través de fórmulas matemáticas que consideran diferencias genéticas individuales14. Una de estas fórmulas para cuantificar la carga de la enfermedad es la desarrollada por Penney et al.15, quienes consideran carga de la enfermedad=edad actual×(CAG-35,5). Esta fórmula ha sido utilizada en varios estudios7,14-16.
Campodonico et al.17 diseñaron una fórmula para predecir la aparición de síntomas motores teniendo en cuenta el tamaño del número de repeticiones de CAG y la edad de inicio de la aparición de la enfermedad en los padres. Brandt et al.18 modificaron la fórmula a 2variables: repeticiones CAG y edad de inicio parental: esta fórmula predice el 62% de la variación del tiempo hasta el inicio de la enfermedad.
En el presente estudio se plantean las siguientes hipótesis: a) los participantes portadores de EH ejecutarán con una mayor variabilidad las tareas cognitivas, a diferencia de los no portadores y b) dependiendo de la carga de la enfermedad y cercanía a la etapa manifiesta (edad de inicio de síntomas), el grupo de portadores tendrá una mayor variabilidad en la ejecución de tareas cognitivas.
MétodoEl Instituto Nacional de Neurología y Neurocirugía Manuel Velasco Suárez, a través del departamento de genética, ofrece a personas en riesgo de heredar EH un Programa de Diagnóstico Predictivo para la Enfermedad de Huntington (PDP-EH)19. Dicho programa está aprobado por los comités de investigación y ética del instituto y se ajusta a las directrices recomendadas por la Asociación Internacional de Huntington, la Federación Mundial de Neurología y el Grupo de Investigación sobre la corea de Huntington20.
En el periodo 2002-2019, un total de 189 personas brindaron su consentimiento informado verbal y por escrito para participar en el PDP-EH. Un equipo multidisciplinario (genetista clínico, psiquiatras, neurólogos, neuropsicólogos, trabajador social) fue responsable de realizar evaluaciones tanto clínicas como cognitivas a todos los participantes. Como criterio de exclusión se consideró la presencia de apatía, ansiedad o depresión; los casos detectados se refirieron a la clínica de psiquiatría y se propuso posponer su participación hasta observar una mejoría psiquiátrica.
Los resultados obtenidos en las diferentes evaluaciones se ingresaron en una base de datos en el programa SPSS versión 22.0. Para cumplir con los objetivos de este estudio se depuraron los datos de dicha base de acuerdo con los criterios de exclusión establecidos: antecedentes de riesgo suicida o cualquier trastorno psiquiátrico no tratado, pruebas neuropsicológicas incompletas, desconocimiento del resultado molecular y síntomas evidentes de la enfermedad o de algún otro padecimiento neurológico. Fueron descartados 30 participantes por no contar con las pruebas cognitivas completas y otros 13 por desconocerse el resultado de la mutación; quedaron un total de 146 participantes, según el resultado molecular.
Para fines prácticos en este trabajo, la muestra se categorizó como en otros estudios7,14,15,18, y se dividió en 2grupos: portadores (PEH) aquellos que tuvieran>36 repetidos CAG del gen de la huntingtina y no portadores (NPEH) quienes tuvieran <36 repeticiones CAG en ambos alelos.
InstrumentosEl PDP-EH se divide en varias sesiones y una de estas se centra en la evaluación neurológica y neuropsicológica de los participantes. En dicha sesión se aplica un cuestionario para obtener datos sociodemográficos y clínicos y se realiza la evaluación neurológica utilizando la escala Unified Huntington's Disease Rating Scale19. La evaluación cognitiva se hizo a través de: Mini-Mental State Examination (MMSE)21 y de pruebas neuropsicológicas estandarizadas en población mexicana22 y enfocadas a valorar funciones ejecutivas, como en otros estudios desarrollados con personas en riesgo de EH23-26. La prueba Stroop color-palabra es una medida de funcionamiento ejecutivo que consta de 3 condiciones: lectura de palabras (Stroop-A), denominación de colores (Stroop-B) e interferencia palabra-color (Stroop-C). Las 2primeras condiciones se han usado en EH para evaluar la velocidad de procesamiento, mientras que la condición de interferencia se ha usado para evaluar funciones ejecutivas, específicamente, inhibición.
El test de símbolos y dígitos (SDMT, por sus siglas en inglés) es una prueba utilizada para evaluar la velocidad de procesamiento.
La fluidez verbal semántica con las categorías «animales» y «frutas» y la fluidez verbal fonológica con las letras «F» y «A» evalúan funciones ejecutivas, en particular, la flexibilidad cognitiva22.
Análisis de datosPara el objetivo de este estudio solo se tienen en cuenta los datos demográficos y cognitivos obtenidos en el PDP-EH. Se realizó un análisis descriptivo de los datos demográficos y se compararon mediante χ2 las variables categóricas y con ANOVA, las continuas. Se analizó la normalidad estadística de los datos cognitivos mediante la prueba de normalidad de Shapiro-Wilk. Los puntajes neuropsicológicos específicos (variables dependientes) se compararon mediante ANOVA de una vía, con el grupo diagnóstico como la variable independiente. Dado el número de comparaciones univariadas, el nivel de significación se estableció en 0,01 para minimizar tanto el error estadístico tipo I como el tipo II.
Para cada participante se determinó el número de repetidos CAG. Se obtuvo el valor de la carga de enfermedad, calculado con la siguiente fórmula: (CAGn− 35,5)×edad. Se obtuvo un rango de 30,00 a 957,00 (M: 292,52; DE: 127,57).
Nosotros usamos la fórmula de Brandt et al.18 para categorizar a los PEH en 3grupos: 1) un grupo denominado «lejos», es decir, lejano al diagnóstico, estimando que tardarán más de 15 años en presentar síntomas cognitivos y ser diagnosticados; 2) un grupo denominado «medio», estimando la presencia de síntomas cognitivos o posible diagnóstico entre 9 y 15 años y 3) un grupo denominado «cerca» con probabilidad de presentar síntomas cognitivos y diagnóstico cercano, estimado en menos de 9 años.
Por último, en el estudio multivariado (MANCOVA) se usaron la carga de enfermedad y la cercanía a la etapa manifiesta (edad de inicio de síntomas) como covariables de las puntuaciones neuropsicológicas. Se reporta la lambada de Wilkins y el tamaño del efecto considerando los siguientes valores (0,04 mínimo; 0,25 moderado; 0,64 grande) en los resultados (η2).
ResultadosDe los 146 participantes, 60 (41,1%) fueron portadores de la expansión (PEH) y 86 (58,9%) no portadores (NPEH). La media del número de repetidos CAG fue 43,95 (DE 3,3; R: 37-53) para los PEH y 20,4 (DE 3,4; R: 15-29) para los NPEH.
Predominaron las mujeres en ambos grupos (56,7% PEH y 61,6% NPEH). La edad media para los PEH fue de 35,9 años y para los NPEH de 38 años. Con respecto a la escolaridad, fue similar el número de años de educación para ambos grupos (PEH 14,5 años; NPEH 14,2 años). No se identificaron diferencias significativas entre los no portadores y los portadores en las variables demográficas.
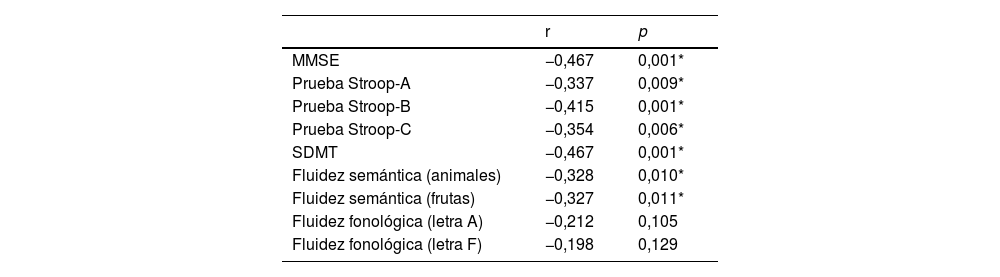
Con respecto a la comparación del desempeño cognitivo entre grupos, se observaron diferencias estadísticamente significativas en las pruebas de MMSE, Stroop-B, SMDT y fluidez fonológica (letra F) (tabla 1).
Desempeño cognitivo de PEH y NPEH
| Grupos | ||||||
|---|---|---|---|---|---|---|
| PEH60 | NPEH86 | |||||
| Variable | M | DE | M | DE | F | p |
| MMSE | 27,8 | 1,8 | 28,8 | 1,2 | 18,358 | 0,001* |
| Prueba Stroop-A | 94,6 | 14,9 | 97,4 | 17,4 | 1,055 | 0,306 |
| Prueba Stroop-B | 60,8 | 13,8 | 68,1 | 10,4 | 13,199 | 0,001* |
| Prueba Stroop-C | 38,1 | 10,1 | 40,9 | 10,1 | 2,600 | 0,109 |
| SDMT | 45,7 | 11,1 | 52,4 | 14,3 | 9,194 | 0,003* |
| Fluidez semántica(animales) | 20,7 | 5,0 | 22,6 | 4,5 | 5,939 | 0,016 |
| Fluidez semántica(frutas) | 14,9 | 3,4 | 15,8 | 3,6 | 2,494 | 0,116 |
| Fluidez fonológica(letra A) | 12,4 | 4,7 | 13,8 | 4,2 | 3,672 | 0,057 |
| Fluidez fonológica(letra F) | 11,8 | 3,8 | 13,6 | 3,5 | 9,515 | 0,002* |
DE: desviación estándar; M: media; MMSE: Mini-Mental State Examination; NPEH: no portador de la enfermedad de Huntington; PEH: portador de enfermedad de Huntington; SDMT: test de símbolos y dígitos.
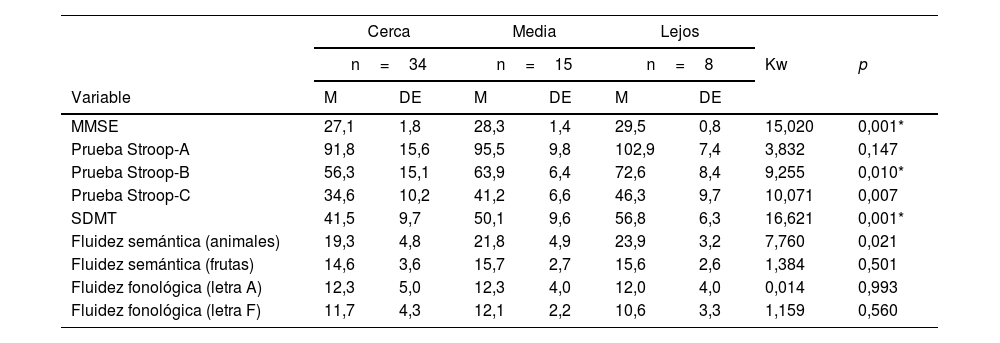
En la tabla 2 se muestran las correlaciones entre la carga de enfermedad y la ejecución neuropsicológica. Las relaciones más fuertes se dan con el MMSE, Stroop (A, B y C), SMDT, fluidez semántica (animales y frutas), donde se observa una correlación inversa entre la carga de enfermedad y las puntuaciones obtenidas en las pruebas neuropsicológicas, lo que indica que, a mayor carga de enfermedad, peor ejecución en la prueba.
Correlación de Pearson entre carga de enfermedad y ejecución neuropsicológica en el grupo de portadores de enfermedad de Huntington
| r | p | |
|---|---|---|
| MMSE | −0,467 | 0,001* |
| Prueba Stroop-A | −0,337 | 0,009* |
| Prueba Stroop-B | −0,415 | 0,001* |
| Prueba Stroop-C | −0,354 | 0,006* |
| SDMT | −0,467 | 0,001* |
| Fluidez semántica (animales) | −0,328 | 0,010* |
| Fluidez semántica (frutas) | −0,327 | 0,011* |
| Fluidez fonológica (letra A) | −0,212 | 0,105 |
| Fluidez fonológica (letra F) | −0,198 | 0,129 |
MMSE: Mini-Mental State Examination; SDMT: test de símbolos y dígitos.
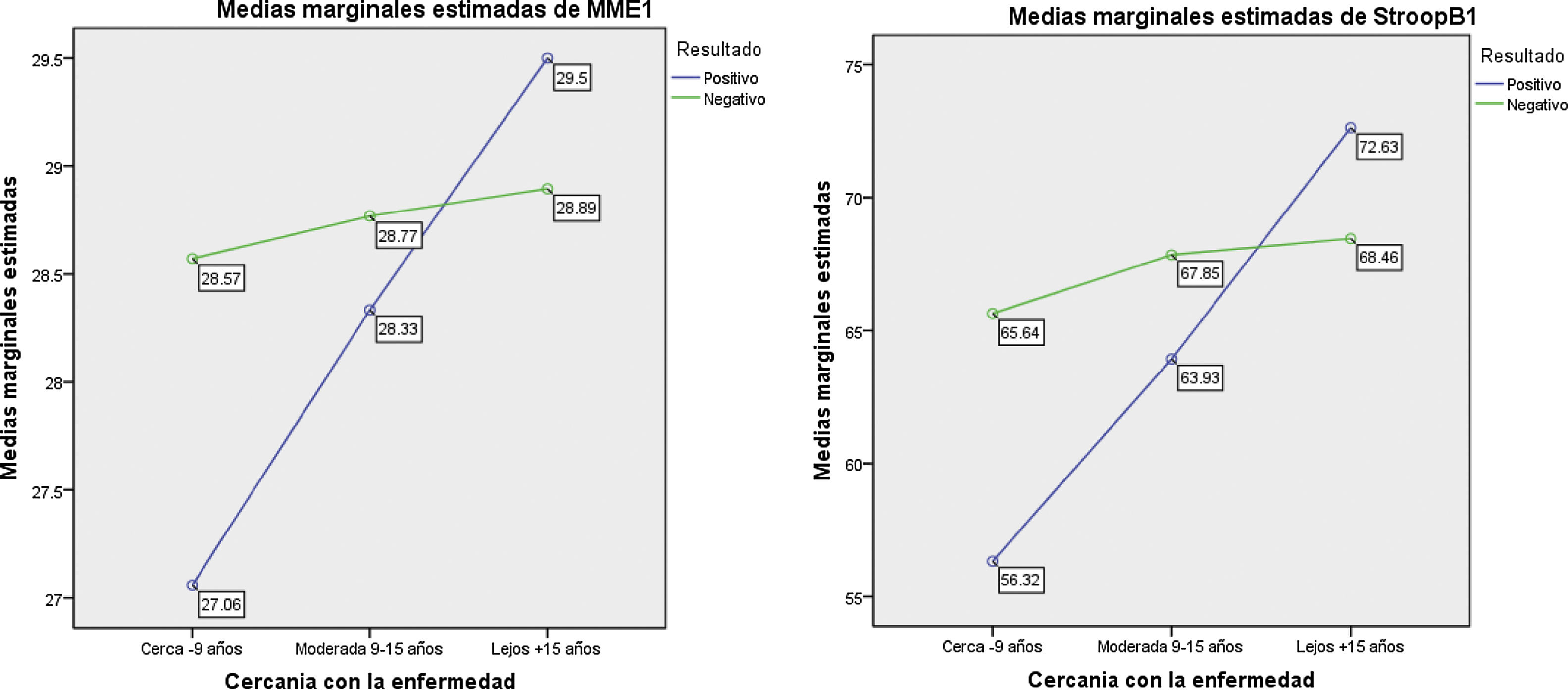
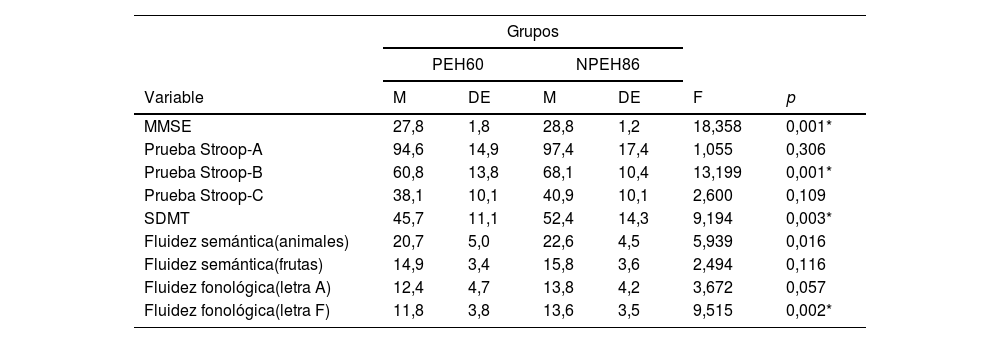
Al categorizar el grupo PEH de acuerdo con la cercanía del diagnóstico, encontramos un grupo lejos (n=8); un grupo medio (n=15) y un grupo cerca (n=34). En 3casos no se contó con la edad de inicio del pariente que desarrolló Huntington, por eso solo se pudieron categorizar 57 de los 60 PEH. Las pruebas de Kruskal-Wallis nos indican los siguientes hallazgos post hoc: para MMSE, el grupo cerca es el que obtiene la puntuación más baja, a diferencia de los otros grupos. Esta misma tendencia se observa para las pruebas de Stroop-B, Stroop-C, SDMT y fluidez semántica (animales) (tabla 3).
Comparación por la probabilidad de inicio cercano de la fase manifiesta (tiempo de diagnóstico motor)
| Cerca | Media | Lejos | ||||||
|---|---|---|---|---|---|---|---|---|
| n=34 | n=15 | n=8 | Kw | p | ||||
| Variable | M | DE | M | DE | M | DE | ||
| MMSE | 27,1 | 1,8 | 28,3 | 1,4 | 29,5 | 0,8 | 15,020 | 0,001* |
| Prueba Stroop-A | 91,8 | 15,6 | 95,5 | 9,8 | 102,9 | 7,4 | 3,832 | 0,147 |
| Prueba Stroop-B | 56,3 | 15,1 | 63,9 | 6,4 | 72,6 | 8,4 | 9,255 | 0,010* |
| Prueba Stroop-C | 34,6 | 10,2 | 41,2 | 6,6 | 46,3 | 9,7 | 10,071 | 0,007 |
| SDMT | 41,5 | 9,7 | 50,1 | 9,6 | 56,8 | 6,3 | 16,621 | 0,001* |
| Fluidez semántica (animales) | 19,3 | 4,8 | 21,8 | 4,9 | 23,9 | 3,2 | 7,760 | 0,021 |
| Fluidez semántica (frutas) | 14,6 | 3,6 | 15,7 | 2,7 | 15,6 | 2,6 | 1,384 | 0,501 |
| Fluidez fonológica (letra A) | 12,3 | 5,0 | 12,3 | 4,0 | 12,0 | 4,0 | 0,014 | 0,993 |
| Fluidez fonológica (letra F) | 11,7 | 4,3 | 12,1 | 2,2 | 10,6 | 3,3 | 1,159 | 0,560 |
Los valores mostrados son medias±desviaciones estándar.
Cerca: estimado en <9 años para el diagnóstico de enfermedad de Huntington; Medio: entre 9 y 15 años para el diagnóstico de enfermedad de Huntington; Lejos: más de 15 años para el diagnóstico de enfermedad de Huntington.
DE: desviación estándar; M: media; MMSE: Mini-Mental State Examination; SDMT: test de símbolos y dígitos.
Para conocer el efecto de la carga de la enfermedad y cercanía (edad) del inicio de los síntomas motores se realizó un análisis multivariado (MANCOVA) para cada variable estadísticamente significativa.
En la figura 1 se observan los resultados del MANCOVA, con las puntuaciones obtenidas en la valoración neuropsicológica. Covariando las puntuaciones de carga de la enfermedad y cercanía del inicio de síntomas, se evidencian diferencias estadísticamente significativas con las puntuaciones neuropsicológicas, lambada de Wilkins (Λ = 0,736; F [5, 135]=3,58; p<0,001), el tamaño del efecto es grande (η2=0,998; r=0,14). El análisis de regresión múltiple (r=0,226; F=4,905; p=0,009) mostró que el efecto MMSE evidencia diferencias estadísticamente significativas entre carga de la enfermedad y cercanía de inicio de síntomas (F(1,135)=−3,116; p=0,002) y el tamaño del efecto fue grande (η2=0,871). Esto significa que cuanto más cerca se encuentran del inicio de síntomas, peor es la ejecución del MMSE entre los pacientes portadores. Por otra parte, se observó una tendencia (r=0,149; F=2,874; p=0,060) que mostró que el efecto Stroop-B evidencia diferencias estadísticamente significativas entre carga de la enfermedad y cercanía de inicio de síntomas (F(1,135)=−2,397; p=0,018), mostrando un tamaño de efecto grande (η2=0,663). La ejecución de los portadores (PEH) fue peor cuanto más cerca se encontraban del inicio de los síntomas, a diferencia de los no portadores (NPEH).
Discusión
Este es el primer estudio en población mexicana de personas que asisten a un programa de diagnóstico predictivo para la EH. Los resultados cognitivos concuerdan con varios resultados previos reportados en la literatura internacional14,23-26.
De acuerdo con los resultados cognitivos, se observa que la media del puntaje en la prueba MMSE de ambos grupos se encuentra en los rangos de funcionamiento cognitivo normal: ningún participante obtuvo menos de 23 puntos, que es el punto de corte para deterioro cognitivo en población hispanohablante27. Varios estudios reportan que tanto el grupo de NPEH como el grupo PEH tienen un funcionamiento cognitivo similar y dentro de rangos normales24,28. Sin embargo, nosotros sí encontramos diferencias estadísticas entre los grupos en el puntaje total del MMSE, además de observar una correlación entre la carga de la enfermedad y el puntaje de esta prueba en el grupo PEH. Estos cambios en el funcionamiento cognitivo global fueron sutiles, pero pueden ser indicador de sintomatología incipiente en el desarrollo de la EH, como señalan Verny et al.23. También se ha encontrado que las puntuaciones en el MMSE se correlacionan con la disminución en el volumen del caudado, lo cual es observable desde etapas prediagnósticas de la EH29.
Por otro lado, se ha descrito que las tareas de atención sostenida, como en el caso de las tareas del Stroop, pueden verse alteradas en estadios presintomáticos de la enfermedad, así como la velocidad de procesamiento, y manifestarse antes de la aparición de síntomas motores en PEH29,30. En este trabajo se observaron diferencias significativas en la ejecución de la tarea Stroop-B, condición de nominación de color: aunque dicha tarea tiene cierto grado de automaticidad, requiere niveles mayores de atención que la tarea de Stroop-A. Se ha observado que las respuestas automáticas, que se ven reflejadas en el rendimiento de la condición de lectura, se deterioran con mayor frecuencia en estadios avanzados de la EH23,29.
Se encontraron diferencias significativas entre grupos y correlaciones con la carga de la enfermedad en la prueba Stroop y SDMT, pruebas utilizadas ampliamente en la valoración de pacientes con EH y que han resultado ser altamente sensibles29,31. En particular, la SDMT ha sido señalada como marcador clínico de tipo cognitivo para evaluar la progresión premanifiesta de la EH32, aunque son tareas en las que influyen más de un proceso cognitivo. En la primera, automatización, atención y velocidad de procesamiento; en la segunda, atención, rastreo visual, velocidad de procesamiento y coordinación visomotora son pruebas que se relacionan, específicamente, con velocidad de procesamiento e inhibición y atención e integración de la información.
Con respecto a la prueba de fluidez verbal, se encontraron diferencias estadísticamente significativas solo en la tarea de fluidez fonológica (letra F), lo cual podría relacionarse con las variaciones en la actividad cognitiva que exige cada una de las pruebas de fluidez verbal. En la mayoría de las investigaciones con PEH no se reporta la categoría o letra utilizada en las diferentes tareas de fluidez verbal24,33,34, lo que hace difícil confirmar si los resultados divergentes en los diferentes estudios se deben al tipo de prueba utilizada. Se sabe que al ser una prueba en la que se otorga un tiempo limitado para responder, la velocidad de procesamiento es un factor importante que impacta en la adecuada ejecución. Por lo tanto, es posible que una deficiencia en la productividad de palabras sea más evidente en las pruebas con mayor disponibilidad léxica, en sujetos en los que además está afectada la velocidad de procesamiento33.
En este sentido, se debe considerar la posibilidad de que las deficiencias en fluidez verbal sean un reflejo de la alteración de la velocidad procesamiento observada en los PEH.
De acuerdo a investigaciones precedentes29,35,36, es posible que las alteraciones cognitivas que se observaron en el grupo PEH deriven de afectaciones de la función del estriado y de los circuitos frontoestriatales, como resultado secundario de la pérdida de las proyecciones del estriado a la corteza cerebral. Cambios en la cognición pueden vincularse a marcadores biológicos antes de la presencia de síntomas motores o disminución funcional. Así, los cambios visomotores y psicomotores pueden aparecer hasta avanzada la enfermedad, mientras que las alteraciones en el funcionamiento ejecutivo se observan más cerca del diagnóstico clínico. Paulsen et al.37 mostraron que los síntomas cognitivos se pueden detectar aproximadamente 15 años antes del inicio motor y tienen una progresión insidiosa, que se hace más rápida cerca del diagnóstico.
De aquí la necesidad de evaluar los aspectos cognitivos en etapas tempranas y, de acuerdo con Stout et al.38, las pruebas neurocognitivas son indicadores clínicos sólidos del proceso de la enfermedad antes de alcanzar criterios para el diagnóstico motor de la EH. Por lo tanto, se propone que el rendimiento neurocognitivo sea evaluado mediante una batería completa de tareas cognitivas diseñadas para maximizar la sensibilidad a los circuitos neuronales frontoestriados. La comprensión del funcionamiento ejecutivo en personas en riesgo de EH agrega información, aparte de las diferencias en el control motor, y probablemente tendrá un valor utilitario en entornos clínicos39.
Por último, de acuerdo con los resultados moleculares obtenidos en este estudio, observamos que el promedio del número de repeticiones CAG fue 43,95 en el grupo PEH, categorizado como fenotipo de penetrancia completa, según Tabrizi et al.4. Se ha observado que la variable tiempo estimado de inicio de la fase manifiesta de EH se relaciona con el desempeño en las pruebas cognitivas; es decir, quienes se encuentran más cerca de esta fase tienen un rendimiento menor que quienes se encuentran lejos, como se pudo corroborar en este trabajo. Se ha demostrado que el «deterioro cognitivo leve» está presente en hasta la mitad de las personas del PEH cercanas al diagnóstico motor40.
ConclusionesDe acuerdo con los resultados obtenidos en el grupo PEH con probabilidad de inicio cercano de la fase manifiesta, se observa un nivel menor de desempeño en comparación con el grupo PEH con probabilidad de inicio lejano de la fase manifiesta, principalmente en la ejecución de pruebas neuropsicológicas que evalúan velocidad de procesamiento y atención. Esto confirma que la disfunción cognitiva, específicamente la función prefrontal, es la alterada de manera más precoz y aparece varios años antes del diagnóstico motor de la EH29.
Pruebas formales tanto cognitivas como motoras que evalúen las habilidades de una persona en riesgo de EH pueden ser de utilidad para identificar la aparición de los diversos síntomas de la enfermedad y para caracterizar su gravedad, además de facilitar estrategias de compensación y estimulación que mejoren la capacidad funcional y la calidad de vida.
Conflictos de interesesLos autores niegan conflictos de intereses de cualquier tipo.

