











Dada la escasez de directrices abordando este tema y con motivo de la futura creación de la Unidad de Cuidados Paliativos (CP) en nuestro centro de neurorrehabilitación, los miembros del equipo médico de la Clínica San Vicente hemos decidido proponer una serie de sugerencias sobre el empleo de fármacos antiepilépticos (FAEs) en el manejo de las crisis epilépticas (CEs) en CP.
MétodosBúsqueda de artículos en PubMed, últimos libros y recomendaciones de las guías de práctica clínica y sociedades científicas publicadas más relevantes, referentes al manejo de las CEs en CP.
ResultadosLa confección de este tipo de guías, además de identificar pacientes candidatos a recibir CP, es fundamental para garantizar un buen control sintomático de las CEs y evitar el sufrimiento innecesario de estos enfermos y sus familiares. Dadas las características de estos pacientes, se recomienda usar FAEs con presentación vía parenteral (preferiblemente intravenosa) y un perfil bajo de interacciones. Diazepam y/o midazolam serían los más idóneos para la fase aguda, y levetiracetam, ácido valproico y/o lacosamida para casos refractarios y/o como tratamiento crónico.
ConclusionesEstas recomendaciones deben considerarse una guía de abordaje integral, debiendo adaptarse a la idiosincrasia de cada caso clínico en particular. Sin embargo, se necesitan ensayos clínicos controlados, aleatorizados, bien diseñados, que incluyan muestras amplias de pacientes subsidiarios de CP, para redactar un documento de consenso que permita recomendar con un mayor nivel de evidencia y de forma generalizada, la utilización adecuada, racional y efectiva de FAEs en este ámbito médico-asistencial de elevada complejidad.
Very little has been written on seizure management in palliative care (PC). Given this situation, and considering the forthcoming setting up of the Palliative Care Unit at our neurorehabilitation centre, the Clínica San Vicente, we decided to establish a series of guidelines on the use of antiepileptic drugs (AEDs) for handling seizures in PC.
MethodsWe conducted a literature search in PubMed to identify articles, recent manuals, and clinical practice guidelines on seizure management in PC published by the most relevant scientific societies.
ResultsClinical practice guidelines are essential to identify patients eligible for PC, manage seizures adequately, and avoid unnecessary distress to these patients and their families. Given the profile of these patients, we recommend choosing AEDs with a low interaction potential and which can be administered by the parenteral route, preferably intravenously. Diazepam and midazolam appear to be the most suitable AEDs during the acute phase whereas levetiracetam, valproic acid, and lacosamide are recommended for refractory cases and long-term treatment.
ConclusionsThese guidelines provide general recommendations that must be adapted to each particular clinical case. Nevertheless, we will require further well-designed randomised controlled clinical trials including large samples of patients eligible for PC to draft a consensus document recommending adequate, rational, and effective use of AEDs, based on a high level of evidence, in this highly complex area of medical care.
El envejecimiento de la población y el creciente número de personas con enfermedades crónico-degenerativas y/o cancerígenas representan un reto importante para los servicios de salud. Muchos de estos enfermos, al final de su vida, sufren un padecimiento intenso y precisan una atención sociosanitaria que implica a todos los estamentos asistenciales. En España se calcula que el 50-60% de las personas que fallecen han recorrido un proceso de deterioro en el último año de su vida. Y se estima que un 8-22% de las hospitalizaciones pueden corresponder a enfermos en este período1-3. Existe, además, en nuestra sociedad, una demanda generalizada de atención centrada en la persona, de calidad y a costes razonables, que permita una vida y una muerte dignas. Esta realidad revela la necesidad de realizar un replanteamiento acerca de los objetivos que debe perseguir la medicina actual que, hasta ahora, se ha enfocado de manera excesiva en un enfoque curativo. Daniel Callahan, en un artículo publicado en el año 2000 en la prestigiosa revista New England Journal of Medicine, ya abogaba por reconocer una muerte en paz, como un objetivo del mismo valor e importancia, que la prolongación de la vida y la lucha contra las enfermedades4. Además, durante el transcurso de su enfermedad, los pacientes con tumores encefálicos presentan déficits neurológicos que pueden ser debidos a los efectos primarios tumorales, las complicaciones sistémicas y/o a efectos adversos del tratamiento oncológico. En general, la neurorrehabilitación (NRHB) en las primeras etapas del proceso neoplásico tiene como objetivo restaurar las funciones cognitivas del paciente después de la terapia antitumoral, mientras que en etapas avanzadas es importante para mantener su independencia y calidad de vida. Asimismo, la NRHB ha demostrado producir un efecto positivo significativo, especialmente en la fase aguda de la enfermedad oncológica, con una ganancia en la funcionalidad del paciente comparable a la obtenida con otros modelos contemplados en otras afecciones neurológicas como el ictus o la lesión cerebral traumática. A pesar de todo, la NRHB en estos pacientes está todavía infrautilizada5. La concepción global de los cuidados paliativos (CP) reconoce que las personas con enfermedades distintas del cáncer, que sean irreversibles, progresivas y con una fase terminal, también pueden beneficiarse de su aplicación, por ejemplo, los pacientes en estadio avanzado con enfermedad pulmonar obstructiva crónica, insuficiencia cardíaca, renal y/o hepática o enfermedades neurológicas (ictus, demencias, enfermedad de Parkinson, esclerosis múltiple, esclerosis lateral amiotrófica, etc.)1–8. No obstante, estas guías se centrarán en los pacientes oncológicos, que merecen una especial consideración por su creciente prevalencia y las interacciones ocasionadas entre el tratamiento antiepiléptico (especialmente los fármacos antiepilépticos [FAEs] clásicos) y los regímenes de fármacos antineoplásicos1–10. Para ello, hemos llevado a cabo una revisión sistemática de la literatura recopilada en los artículos, libros y guías de práctica clínica (GPC) más reseñables, publicados en los últimos 16 años (tomando como referencia la primera GPC sobre CP, de 2008, del Plan Nacional para el Sistema Nacional de Salud [SNS] del Ministerio de Sanidad, Servicios Sociales e Igualdad, hasta 2016, año en que ven la luz la última guía oficial de la Sociedad Española de Neurología de práctica clínica en epilepsia y una revisión sistemática realizada por Sauro et al. sobre el estado actual de las guías en epilepsia10), para tratar de esgrimir un modelo de propuesta apropiado, acerca del manejo de las crisis epilépticas (CEs) en CP. En lo que respecta a la clasificación de las evidencias científicas, nos adecuaremos a las recomendaciones revisadas de la European Federation of Neurological Societies del año 2004 (tabla 1)11.
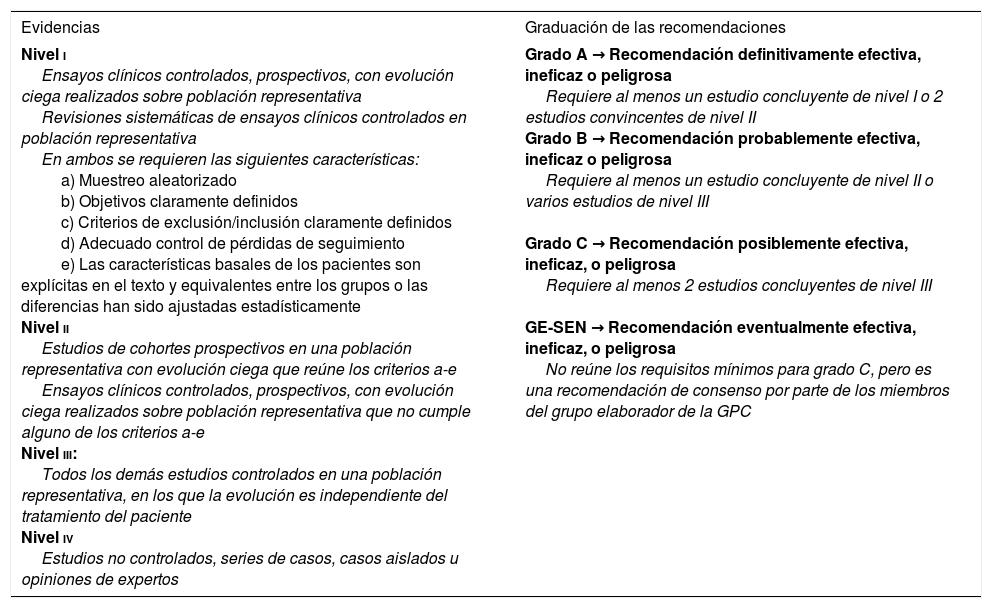
Clasificación del nivel de evidencia para actuaciones terapéuticas
| Evidencias | Graduación de las recomendaciones |
|---|---|
| Nivel i Ensayos clínicos controlados, prospectivos, con evolución ciega realizados sobre población representativa Revisiones sistemáticas de ensayos clínicos controlados en población representativa En ambos se requieren las siguientes características: a) Muestreo aleatorizado b) Objetivos claramente definidos c) Criterios de exclusión/inclusión claramente definidos d) Adecuado control de pérdidas de seguimiento e) Las características basales de los pacientes son explícitas en el texto y equivalentes entre los grupos o las diferencias han sido ajustadas estadísticamente Nivel ii Estudios de cohortes prospectivos en una población representativa con evolución ciega que reúne los criterios a-e Ensayos clínicos controlados, prospectivos, con evolución ciega realizados sobre población representativa que no cumple alguno de los criterios a-e Nivel iii: Todos los demás estudios controlados en una población representativa, en los que la evolución es independiente del tratamiento del paciente Nivel iv Estudios no controlados, series de casos, casos aislados u opiniones de expertos | Grado A → Recomendación definitivamente efectiva, ineficaz o peligrosa Requiere al menos un estudio concluyente de nivel I o 2 estudios convincentes de nivel II Grado B → Recomendación probablemente efectiva, ineficaz o peligrosa Requiere al menos un estudio concluyente de nivel II o varios estudios de nivel III Grado C → Recomendación posiblemente efectiva, ineficaz, o peligrosa Requiere al menos 2 estudios concluyentes de nivel III GE-SEN → Recomendación eventualmente efectiva, ineficaz, o peligrosa No reúne los requisitos mínimos para grado C, pero es una recomendación de consenso por parte de los miembros del grupo elaborador de la GPC |
GE-SEN: Grupo de Estudio de Epilepsia de la Sociedad Española de Neurología; GPC: guía de práctica clínica.
Tomada con permiso de Mercadé Cerdá et al.51, 2016.
La Organización Mundial de la Salud define los CP como «el enfoque que mejora la calidad de vida de pacientes y familias que se enfrentan a los problemas asociados con enfermedades amenazantes para la vida, a través de la prevención y el alivio del sufrimiento, por medio de la identificación temprana y la impecable evaluación y tratamiento del dolor y otros problemas físicos, psicosociales y espirituales». Considera que el equipo sociosanitario debe aproximarse a los enfermos y a sus familiares con el objetivo de responder a sus necesidades, y enumera las siguientes características de los CP1,2: a) proporcionan el alivio del dolor y de otros síntomas que producen sufrimiento; b) promocionan la vida y consideran la muerte como un proceso natural; c)) no se proponen acelerar el proceso de morir ni retrasarlo; d) integran los aspectos psicosociales y espirituales en los cuidados del paciente; e) tienen en cuenta el soporte y los recursos necesarios para ayudar a los pacientes a vivir de la manera más activa posible hasta su muerte; f) ofrecen apoyo a los familiares y a los allegados durante la enfermedad y el duelo; g) mejoran la calidad de vida del paciente; h) se aplican desde las fases tempranas de la enfermedad junto con otras terapias dirigidas a prolongar la vida (como la quimioterapia [QT], la radioterapia [RT], etc.) (fig. 1), e i) incluyen también las investigaciones necesarias para comprender mejor y manejar situaciones clínicas complejas.
 se inician desde el diagnóstico de una enfermedad que amenaza la vida, a la par que los tratamientos curativos. De la misma forma, incluso en las fases finales de la enfermedad, en las que el tratamiento es predominantemente paliativo, puede existir un espacio para el intento destinado a las medidas curativas. Al inicio de la fase final de la vida o situación de enfermedad terminal (SET), el tratamiento curativo ha terminado y los CPs se convierten en cuidados de la SET. Por último, el duelo puede requerir atención especializada durante una fase prolongada. Fuente: Grupo de Trabajo de la Guía de Práctica Clínica sobre Cuidados Paliativos1. Imagen adaptada con permiso de Koekkoek et al.2, 2016.")
Esquema iconográfico, cronológico y conceptual de los cuidados paliativos.
Los cuidados paliativos (CPs) se inician desde el diagnóstico de una enfermedad que amenaza la vida, a la par que los tratamientos curativos. De la misma forma, incluso en las fases finales de la enfermedad, en las que el tratamiento es predominantemente paliativo, puede existir un espacio para el intento destinado a las medidas curativas. Al inicio de la fase final de la vida o situación de enfermedad terminal (SET), el tratamiento curativo ha terminado y los CPs se convierten en cuidados de la SET. Por último, el duelo puede requerir atención especializada durante una fase prolongada.
Fuente: Grupo de Trabajo de la Guía de Práctica Clínica sobre Cuidados Paliativos1.
Imagen adaptada con permiso de Koekkoek et al.2, 2016.
A. Situación de enfermedad terminal (SET): es aquella en la que existe una enfermedad avanzada, incurable, progresiva, sin posibilidades razonables de respuesta al tratamiento específico, que genera problemas como la existencia de síntomas multifactoriales, intensos y cambiantes, originando un gran impacto emocional en enfermos, familiares y equipos terapéuticos, con un pronóstico vital limitado, que provoca una gran demanda de atención, y en la que el objetivo fundamental consiste en la promoción del bienestar y la calidad de vida del enfermo y de la familia, basada en el control de síntomas, el soporte emocional y la comunicación (Plan Nacional de Cuidados Paliativos, bases para su desarrollo)8.
B. Paciente paliativo: es aquel que presenta una enfermedad avanzada progresiva e incurable, en el que el tratamiento específico para la enfermedad ha sido optimizado al máximo, que sufre múltiples problemas y/o síntomas intensos que no mejoran a pesar del tratamiento adecuado, y cuya vida es limitada. Estos pacientes pueden ser oncológicos o no oncológicos6,8 (tabla 2).
Criterios de SET en el paciente oncológico y no oncológico6,8
| Paciente oncológico (en el cual nos centraremos en esta guía) | Paciente no oncológico |
|---|---|
| – Presencia de una enfermedad oncológica avanzada, progresiva e incurable; con diagnóstico histológico demostrado, tras haber recibido terapéutica estándar eficaz. En algunas situaciones especiales, y de manera excepcional, se aceptará la ausencia de diagnóstico histológico. En estas situaciones, en las que por la situación clínica del paciente no se considera adecuado proceder a una investigación exhaustiva de su neoplasia, se deberá haber excluido tumores potencialmente tratables – Escasa o nula posibilidad de respuesta al tratamiento activo específico para su patología oncológica. En determinados escenarios se deben utilizar recursos considerados como específicos por su impacto favorable sobre la calidad de vida (QT oral, RT, hormonoterapia, bisfosfonatos, moléculas en 3.ª y/o 4.ª línea, etc.) – Presencia de problemas o síntomas intensos, múltiples, multicausales y cambiantes Impacto emocional en el paciente, familia y equipo terapéutico, relacionado con el proceso de morir – Pronóstico vital limitado a los últimos meses de vida según criterio del especialista correspondiente, a excepción de aquellas situaciones clínicas complejas que aconsejen CP por la previsión de ganancia en calidad de vida | – Presencia de una enfermedad avanzada, progresiva, sin respuesta al tratamiento médico y/o quirúrgico – El tratamiento específico para la patología de base ha sido optimizado al máximo posible para el paciente. En general, el tratamiento específico, cuando existe, debe mantenerse en la fase final de la enfermedad. La sustitución del tratamiento específico por tratamiento paliativo puro es una excepción justificada solo en situaciones de proximidad del fallecimiento – Presencia de problemas o síntomas intensos, múltiples, multifactoriales y cambiantes a pesar del empleo del adecuado tratamiento específico, precisando varias visitas a urgencias, hospitalizaciones, etc. en los últimos 6 meses – Impacto emocional en el paciente, familia y equipo terapéutico, relacionado con la presencia, explícita o no, de la muerte, con numerosas demandas de atención sanitaria en domicilio, residencias asistidas, etc. – Pronóstico vital limitado: los pacientes susceptibles de una atención específica son aquellos que presenten una situación más cercana al final de la vida. Los instrumentos que pueden facilitar la discriminación adecuada en la mayoría de las enfermedades de base se basan en el diagnóstico y el pronóstico |
| Criterios de SET en determinadas patologías no oncológicas | |
| – ICC: síntomas de ICC en reposo a pesar del tratamiento (incluyendo al menos un IECA y un diurético), ICC grado iv con FEVI ≤ 20%. Arritmias no controlables a pesar del tratamiento, historia de síncopes y/o disnea severa – Insuficiencia respiratoria crónica y EPOC: EPOC muy grave (estadio iv) con FEVI < 30% o FEVI < 50% en presencia de cor pulmonale o fallo cardíaco derecho, hipoxemia en reposo con O2 domiciliario (pO2 ≤ 55mmHg o saturación parcial de O2 ≤ 88%), hipercapnia (pCO2 > 45 mmHg), pérdida de peso ≥ 10% en los últimos 6 meses y/o taquicardia en reposo ≥ 100 lpm – Insuficiencia hepática: enfermo no candidato a trasplante hepático, ascitis refractaria a restricción de líquidos y diuréticos, peritonitis bacteriana, síndrome hepatorrenal, encefalopatía hepática refractaria a restricción proteica, lactulosa y neomicina, sangrado recurrente por varices esofágicas a pesar de trasplante hepático adecuado, pérdida de peso progresiva y/o malnutrición – Insuficiencia renal: pacientes que podrían ser candidatos a diálisis pero rechazan la misma o el trasplante renal, tienen una expectativa de vida menor de 6 meses, presentan oliguria < 400 ml/24 h, pericarditis urémica y/o síndrome hepatorrenal – Demencia avanzada: deterioro cognitivo muy severo, donde el paciente es incapaz de comunicarse verbalmente con sentido, de reconocer a sus cuidadores, etc., con complicaciones médicas en el último año como neumonía por aspiración, infecciones del tracto urinario, sepsis, fiebre recurrente tras recibir antibioterapia y/o dificultad para llevar a cabo la deglución – Esclerosis lateral amiotrófica: su tratamiento es siempre paliativo, ya que se produce la muerte de neuronas motoras, sin afectar a neuronas sensitivas, musculatura ocular, esfínteres y/o función cognitiva (estos pacientes reúnen siempre criterios, independientemente del grado de afectación) |
CP: cuidados paliativos; EPOC: enfermedad pulmonar obstructiva crónica; FEVI: fracción de eyección del ventrículo izquierdo; ICC: insuficiencia cardíaca crónica; IECA: inhibidor de la enzima conversora de angiotensina; SET: situación de enfermedad terminal.
Epilepsia y neoplasias1–104 La epilepsia, conocida como enfermedad comicial (por ser un signo de mal presagio en los comicios romanos), afecta al 0,5-1% de la población, con 2 picos de incidencia: la infancia y la senectud26. A su vez, las CEs no son inusuales en CP, sobre todo en pacientes con neoplasias encefálicas, ya que por ejemplo en el caso de los gliomas (los tumores cerebrales primarios más frecuentes a nivel cerebral) hasta en el 88%87,100 de los casos aparecerán a lo largo de la evolución tumoral5,78–89. Su origen parece multifactorial, con una gran contribución del tejido neuronal sano peritumoral78,90. El control de las CEs es crucial en el manejo de estos enfermos y un motivo frecuente de consulta. Este impacto sanitario justifica el afrontamiento diagnóstico-terapéutico mediante una protocolización asistencial correcta y efectiva en el plazo temporal más breve posible. Debiendo tener en todo momento claro que no toda convulsión es una CE y no todas las CEs presentan convulsiones55. Hasta un 4% de los pacientes oncológicos tienen CEs provocadas por otras causas capaces de inducir la aparición de CEs57–64 (tabla 3).
Aproximación etiológica de las CEs en pacientes con cáncer
| Relacionadas con la afectación del SNC |
| — Tumor cerebral primario — Metástasis cerebrales — Enfermedad cerebrovascular — Síndrome de leucoencefalopatía posterior reversible — Meningoencefalitis — Metástasis leptomeníngeas, etc. |
| Relacionadas con el tratamiento |
| — Quimioterapia: citarabina, metotrexato, cisplatino, bevacizumab, etopósido, interferón alfa, ifosfamida, ciclofosfamida, L-asparaginasa, vincristina, interleucina-2, nitrosoureas (carmustina, lomustina), antraciclinas (doxorrubicina), etc. — Tóxico/metabólico: insuficiencia renal, insuficiencia hepática, síndrome de lisis tumoral, púrpura trombótica trombocitopénica, alteraciones hidroelectrolíticas, hipoglucemia, hipoxia/embolismo pulmonar, etc. — Otros fármacos: meperidina, neurolépticos, bisfosfonatos, ondansetrón, imipenem, etc. — Radioterapia craneal (encefalopatía aguda por radiación, radionecrosis diferida del lóbulo temporal, etc.) |
CEs: crisis epilépticas.
Adaptada con permiso de Corredera García y Becerra Cuñat57, 2012.
Etiología1–106 Los tumores del sistema nervioso central (SNC) son la causa más frecuente de epilepsia en el intervalo etario de 41-60 años26. A su vez, las CEs son una complicación habitual y grave en el paciente oncológico. Su causa más frecuente son los tumores intracraneales52, muy corrientes en el caso del tumor disembrioplásico neuroepitelial (100%), el ganglioglioma (80-90%), los primarios de estirpe glial, especialmente de bajo grado (75%)92,93 (favoreciendo la epileptogénesis mediante la liberación astrocitaria de glutamato)78,90, los meningiomas (22-60%), el glioblastoma multiforme (29-49%), las metástasis encefálicas (20-35%), el tumor leptomeníngeo (10-15%) y el linfoma cerebral primario del SNC (10%); siendo hasta en el 45%100 su primera manifestación semiológica81,82,101 (más común si existen lesiones múltiples y/o histología de melanoma)65. El crecimiento induce inicialmente signos focales y en más del 80%, se detectan con posterioridad al diagnóstico del tumor primario (metástasis metacrónicas). Menos frecuente es que se diagnostiquen como primera manifestación de la enfermedad (metástasis sincrónicas)65. Como se ha introducido anteriormente, las CEs pueden deberse a diversos factores1 (tabla 2): a) tumores cerebrales primarios y metástasis cerebrales: sobre todo de cáncer de pulmón, de mama y melanoma (siendo con más frecuencia múltiples en este último caso); poco probables en tumores de próstata, orofaringe o piel65; b) QT: especialmente si es a dosis altas y/o en caso de insuficiencia hepática y/o renal; c) trastornos metabólicos: unas veces generados por el propio tumor (hipercalcemia en cánceres de pulmón, próstata, mama y en el mieloma múltiple), y otras por los fármacos (hiponatremia por ciclofosfamida, hipocalcemia por bisfosfonatos, hipomagnesemia por cisplatino, etc.); d) fármacos (véase la tabla 3); e) síndromes paraneoplásicos; f) complicaciones cerebrovasculares (ictus, trombosis de senos venosos, etc.; en una serie de 96 pacientes con ictus y cáncer del Memorial Sloan-Kettering Cancer Center de Nueva York, un 8% presentó CEs76); g) infecciones del SNC (herpéticas, mayoritariamente); h) síndrome de inmunodeficiencia adquirida (sida) (criptococosis y toxoplasmosis de topografía encefálica, síndrome de encefalopatía aguda fulminante, etc.)105,106, e i) RT craneal (véase la tabla 3).
Clínica1–78 Pueden presentarse diferentes tipos de CEs y llegar al status epileptitus (SE). Esto depende de la velocidad de crecimiento lesional y su circunscripción, siendo la localización temporal, frontal y parietal las que más frecuentemente producen CEs focales52. El SE es infrecuente en los pacientes con tumores cerebrales, pero asocia una mortalidad del 6-35%78.
Definiciones y clasificaciones de la International League Against Epilepsy (ILAE) 2010 y 20157–51,107–116A. Convulsiones: movimientos involuntarios, generalmente mantenidos (tónicos) o interrumpidos (clónicos), consecuencia de una alteración en el funcionamiento encefálico, caracterizada por una descarga anormal, hipersincrónica y autolimitada de un conjunto de neuronas corticales del SNC.
B. CEs: manifestación clínica derivada de dicha descarga, hemisférica localizada en las CEs focales (parciales), o bihemisférica en las CEs generalizadas. La expresión clínica de cualquier CE puede incluir alteraciones del nivel de consciencia y/o manifestaciones motoras, sensitivas, autonómicas y/o psíquicas, percibidas por el paciente o, habitualmente, por observadores externos. Por lo general son episodios estereotipados, paroxísticos, breves y transitorios o autolimitados32,44.
C. SE: es la emergencia neurológica más frecuente tras el ictus38, de importancia vital ya que comporta una alta tasa de morbimortalidad, y su pronóstico se establece en términos de supervivencia23. Es una afección caracterizada por una CE suficientemente prolongada o CEs subintrantes a intervalos suficientemente breves, sin llegar a recuperar completamente el nivel de consciencia entre ellas, para crear una condición epiléptica invariable y duradera32,44. Existen 2 subtipos principales de SE: 1) convulsivo (SEC): actividad epiléptica caracterizada por bien: a) una CE continua ≥ 5 min (si SE tónico-clónico), ≥ 10 min (si SE focal con alteración del nivel de consciencia) o ≥ 10-15 min (SE de ausencias); b) ≥ 2 CEs sin recuperación completa de la consciencia entre las mismas; c) o CEs en salvas o acúmulos (seizure clusters) (≥ 3 CEs convulsivas en 24 h)32–44,51,111–116; y 2) no convulsivo (SENC): CEs sin actividad motora reconocible (o predominante) y con trazado electroencefalográfico crítico continuo. Habitualmente, se manifiesta en la clínica con un descenso del nivel de consciencia.
D. SE refractario (SER): SE que persiste a pesar del tratamiento con 2 FAEs (primera y segunda línea) indicados, a dosis adecuadas, y/o actividad epiléptica ≥ 30 min. Se presentan en el 31-43% de los pacientes con SE, casi la mitad de los cuales precisa un coma anestésico para su control y conlleva una tasa de mortalidad de hasta el 39%107.
E. SE superrefractario (SERR): si actividad epiléptica ≥ 24 h a pesar del tratamiento antiepiléptico.
F. Epilepsia: afección crónica en la que el individuo está predispuesto a presentar CEs recurrentes (≥ 2 CEs ó 1 CE si se objetiva una lesión estructural mediante pruebas complementarias, neuroimagen y/o electroencefalograma [EEG]), con consecuencias cognitivas y/o psicosociales.
G) Epilepsia refractaria (ER): tiene lugar cuando no se obtiene una evolución libre de CEs después probar al menos 2 FAEs, en monoterapia o asociados, siempre que sean apropiados al tipo de epilepsia, administrados de forma adecuada y no retirados por intolerancia. Se entiende por evolución libre de CEs la ausencia de cualquier tipo de CE durante un período mínimo superior a 3 veces el tiempo intercrisis en el año previo al tratamiento o bien durante un año (siendo válido el período mayor de ambos)14,15. Aparece en el 12-50% de los pacientes con neoplasias encefálicas, especialmente en tumores de bajo grado, invocándose la posible implicación causal de genes de multirresistencia a fármacos (multidrug resistance [MDR] genes)82.
Desde un punto de vista práctico, y de cara al tratamiento, conviene clasificar las CEs según los aspectos clínicos y etiológicos27–74: a) Clasificación de la liga internacional contra la epilepsia 2010 (International League Against Epilepsy [ILAE]) (tabla 4)16,17,27–30,48,49,101; b) clasificación etiológica de las CEs (tabla 5)27,31; c) clasificación de los SE 201526–44: teóricamente hay tantos tipos de SE como tipos de CEs. En la práctica clínica, se habla de SE o SENC (tablas 6 y 7).
Clasificación de los tipos de CEs según la International League Against Epilepsy (ILAE) de 201016–18,27–30,50,51,101
| CEs focales (antiguas parciales) (activación inicial de un conjunto de redes neuronales limitado a una parte localizada o más amplia de un hemisferio cerebral) | CEs generalizadas (la descarga epiléptica inicial afecta a ambos hemisferios simultáneamente. También se acepta que la descarga epiléptica se origina en algún punto, dentro de redes neuronales bilateralmente distribuidas a nivel cortical o subcortical pero no necesariamente todo el córtex cerebral, y difunde rápidamente) |
|---|---|
| Sin alteración del nivel de consciencia (antigua CE parcial simple-epilepsia rolándica) — Con componentes objetivables a nivel motor (fenómenos miocolónicos, clónicos, tónicos, versivos óculo-cefálicos-tronculares, de espasmos, atónicos, mioclónicos negativos, acinéticos y/o hipomotores/de inhibición conductual) o autonómico (fenómenos de horripilación, palidez, taquicardia, náuseas y/o vómitos) – Englobando fenómenos subjetivos de índole psíquica o sensorial (fenómenos a nivel somatosensorial, visual, auditivo, olfativo, gustativo y/o nociceptivo), correspondiéndose con el concepto de «aura» Con alteración del nivel de consciencia o discognitiva (antigua CE parcial compleja-epilepsia temporal): tipo de CE más frecuente de comienzo en la edad adulta. El paciente no responde al estímulo aplicado y se desencadenan fenómenos automotores o automatismos (oroalimentarios, miméticos, carpo-pedales, gestuales, dacrísticos, vocales, hipercinéticos/hipermotores, etc.) — Con evolución a una CE bilateral convulsiva (antigua parcial secundariamente generalizada): comprendiendo los componentes tónico, clónico y/o tónico-clónicoa | Tónico-clónicas (antiguo grand mal, tipo de CE más frecuente secundaria a trastornos metabólicos): en un 15% comienzan con aura motora (p. ej., desviación oculocefálica), y posteriormente se siguen de la fase tónico-clónica (espasmo flexor tónico, seguido de extensión tónica con cierre brusco de la boca, espiración forzada, apnea, cianosis y signos autonómicos, que incluyen aumento de frecuencia cardíaca, presión arterial y presión intravesical, disminución del tono esfinteriano, con incontinencia urinaria, piloerección, enrojecimiento o cianosis facial y/o midriasis binocular). Después, se observa una fase vibratoria hasta llegar a la fase clónica y, por último, la fase postictal con hipotonía, relajación de esfínteres y recuperación progresiva del nivel de consciencia (< 30min). EEG ictal: ritmos rápidos en fases tónicas, con OLs en la fase clónica. — En cualquier combinación (mioclónica, mioclónica-atónica y/o mioclónica-tónica) Ausencias — Típicas (antiguo petit mal): fenómeno paroxístico compuesto por desconexión del medio, de inicio y fin bruscos (sin aura y con recuperación inmediata), con una duración breve. Caracterizada por una interrupción en la actividad que el paciente realiza, sin caídas ni fenómenos motores significativos y con alteraciones típicas en el EEG. EEG ictal: P-O a 3 ciclos/s (Hz) — Atípicas: desconexión del medio de inicio y fin más graduales y duración mayor. EEG ictal: P-O irregular (2-2,5 Hz) — Con características especiales • Ausencias mioclónicas • Mioclonías palpebrales Mioclónicas: movimientos involuntarios, arrítmicos y rápidos, con diferentes localizaciones (únicas o múltiples; axiales, proximales o distales) e intensidades (desde imperceptibles a masivas con caídas y traumatismos). EEG ictal: PP-O o puntas y/u ondas agudas: — Mioclónicas simples — Mioclónicas atónicas (antiguas CEs mioclónicas astáticas) — Mioclónicas tónicas Clónicas: sacudidas musculares rítmicas, de pequeña extensión, sin efecto locomotor. EEG ictal: actividad rápida (10 Hz) y ondas lentas. Tónicas: rigidez muscular brusca mantenida, principalmente en extremidades superiores. EEG: actividad rápida de bajo voltaje o ritmos rápidos (9-10 Hz). Atónicas (antiguos drop-attacks o CEs astáticas): se produce disminución brusca del tono muscular flexor y extensor del cuello, tronco y extremidades. Como consecuencia, se producen caídas sobre glúteos, propulsión o retropulsión o simples cabeceos. EEG ictal: P-O lenta o depresión del trazado electroencefalográfico y/o con desincronización |
En otro apartado se englobarían las CEs inclasificables que comprenden todas las CEs que no se pueden clasificar debido a datos incompletos y/o inadecuados y algunas que no coinciden con la clasificación descrita. Incluyen algunas CEs neonatales (p. ej., movimientos oculares rítmicos, de masticación, de natación, etc.), según la nomenclatura de la clasificación de la ILAE de 1981, y espasmos epilépticos según la de 2010. Por otro lado, en la clasificación de 2010 del grupo de Berg se enfatiza en la importancia de dar descripciones precisas de las CEs, atendiendo a sus manifestaciones motoras, cognitivas, autonómicas y/o senso-experienciales. Cuando estas ocurren de forma secuencial, el orden de dicha sucesión debe procurar quedar también descrito. Por otro lado, las CEs focales motoras son el tipo de CE predominante en los pacientes con metástasis cerebrales90.
CEs: crisis epilépticas; EEG: electroencefalograma; OLs: ondas lentas; P-O: punta-onda; PP-O: polipunta-onda.
Según la última propuesta operativa de la ILAE 2016 podría existir una progresión hacia una CE «bilateral tónico-clónica»30.
Clasificación del Colegio Americano de Médicos de Emergencias (American College of Emergency Physicians [ACEP]) de las CEs según etiología22,27,31
| CEs provocadas | CEs no provocadas o espontáneas | |
|---|---|---|
| CSA | CSR | Etiología indeterminada |
| CSA con lesión agudaa – Postraumática (TCE, cirugía) en la primera semana – Ictus hemorrágico > isquémico en la primera semana – Infección del SNC en la primera semana | CEs con lesión subaguda/crónicaa – Malformaciones, lesiones congénitas y perinatales – Ictus evolucionados – Lesiones cerebrales residuales (cicatricial, gliosis, malacia) – LOEs cerebrales – Cambios degenerativos | CEs idiopáticasb – Antecedentes personales – Adecuación con la edad – Factores favorecedores (↓ umbral epileptógeno) |
| CSA tóxico-metabólicasb – Fármacos y tóxicos – Desequilibro metabólico | CEs criptogénicasa – Probable sintomática – Ampliar estudio etiológico | |
CEs: crisis epilépticas; CSA: crisis sintomáticas agudas; CSR: crisis sintomáticas remotas; LOEs: lesiones ocupantes de espacio; SNC: sistema nervioso central; TCE: traumatismo craneoencefálico.
Adaptada con permiso de Fernández Alonso, 201327.
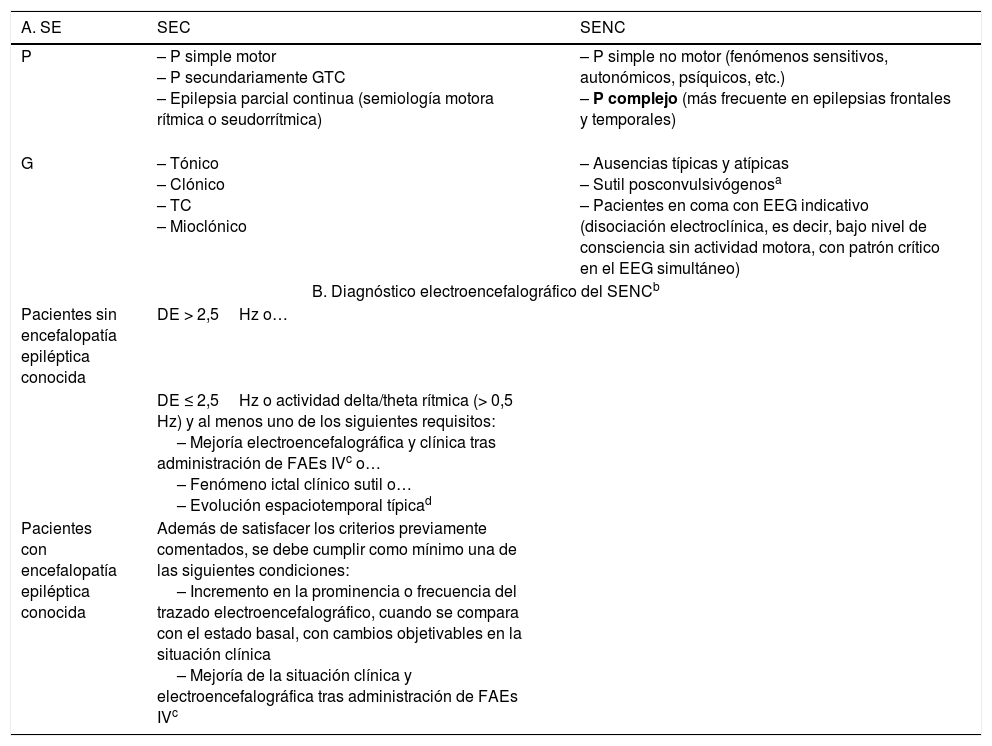
A. Clasificación clásica del SE clásica19,27–37,51. B. Criterios de consenso electroencefalográficos de Salzburgo para el diagnóstico del SENC
| A. SE | SEC | SENC |
|---|---|---|
| P | – P simple motor – P secundariamente GTC – Epilepsia parcial continua (semiología motora rítmica o seudorrítmica) | – P simple no motor (fenómenos sensitivos, autonómicos, psíquicos, etc.) – P complejo (más frecuente en epilepsias frontales y temporales) |
| G | – Tónico – Clónico – TC – Mioclónico | – Ausencias típicas y atípicas – Sutil posconvulsivógenosa – Pacientes en coma con EEG indicativo (disociación electroclínica, es decir, bajo nivel de consciencia sin actividad motora, con patrón crítico en el EEG simultáneo) |
| B. Diagnóstico electroencefalográfico del SENCb | ||
| Pacientes sin encefalopatía epiléptica conocida | DE > 2,5Hz o… | |
| DE ≤ 2,5Hz o actividad delta/theta rítmica (> 0,5 Hz) y al menos uno de los siguientes requisitos: – Mejoría electroencefalográfica y clínica tras administración de FAEs IVc o… – Fenómeno ictal clínico sutil o… – Evolución espaciotemporal típicad | ||
| Pacientes con encefalopatía epiléptica conocida | Además de satisfacer los criterios previamente comentados, se debe cumplir como mínimo una de las siguientes condiciones: – Incremento en la prominencia o frecuencia del trazado electroencefalográfico, cuando se compara con el estado basal, con cambios objetivables en la situación clínica – Mejoría de la situación clínica y electroencefalográfica tras administración de FAEs IVc | |
DE: descargas epileptiformes (puntas, polipuntas, puntas-ondas, complejos punta-onda lenta); EEG: electroencefalograma; FAEs iv: fármacos antiepilépticos por vía intravenosa. G: generalizado; P: parcial; SE: status epilepticus; SEC: SE convulsivo; SENC: SE no convulsivo; TC: tónico-clónico.
SENC generalizado sutil: actividad motora ligera, que aparece a continuación de la actividad epileptiforme ictal tras el cese de la actividad motora evidente tras un SEC generalizado infratratado o no tratado. Compuesta por signos motores menores (clonías faciales y/o distales, nistagmo, desviación ocular, etc.). Suelen ser pacientes gravemente enfermos, ingresados en UCI con un nivel de consciencia severamente disminuido (generalmente en coma), en el contexto de un daño cerebral focal, con una mortalidad que puede llegar a alcanzar el 65%40. Por ello, resulta fundamental para su identificación, conseguir la apertura ocular del paciente39,40.
Adaptada con permiso de Fernández Alonso, 201327.
b El diagnóstico del SENC debe basarse en la conjunción de los hallazgos clínicos y electroencefalográficos. La semiología clínica que debe alertar sobre un posible SENC es necesario que perdure ≥ 10 min.
Si la mejoría electroencefalográfica no asocia una mejoría clínica o si es fluctuante sin una evolución definida, debería considerarse como un posible SENC.
Inicio incremental (aumento en el voltaje y cambio en la frecuencia), evolución en el patrón electroencefalográfico (cambio en la frecuencia > 1Hz o en la localización), o finalización decreciente (en voltaje o frecuencia).
Tomada con permiso de Trinka y Kälviäinen, 201644.
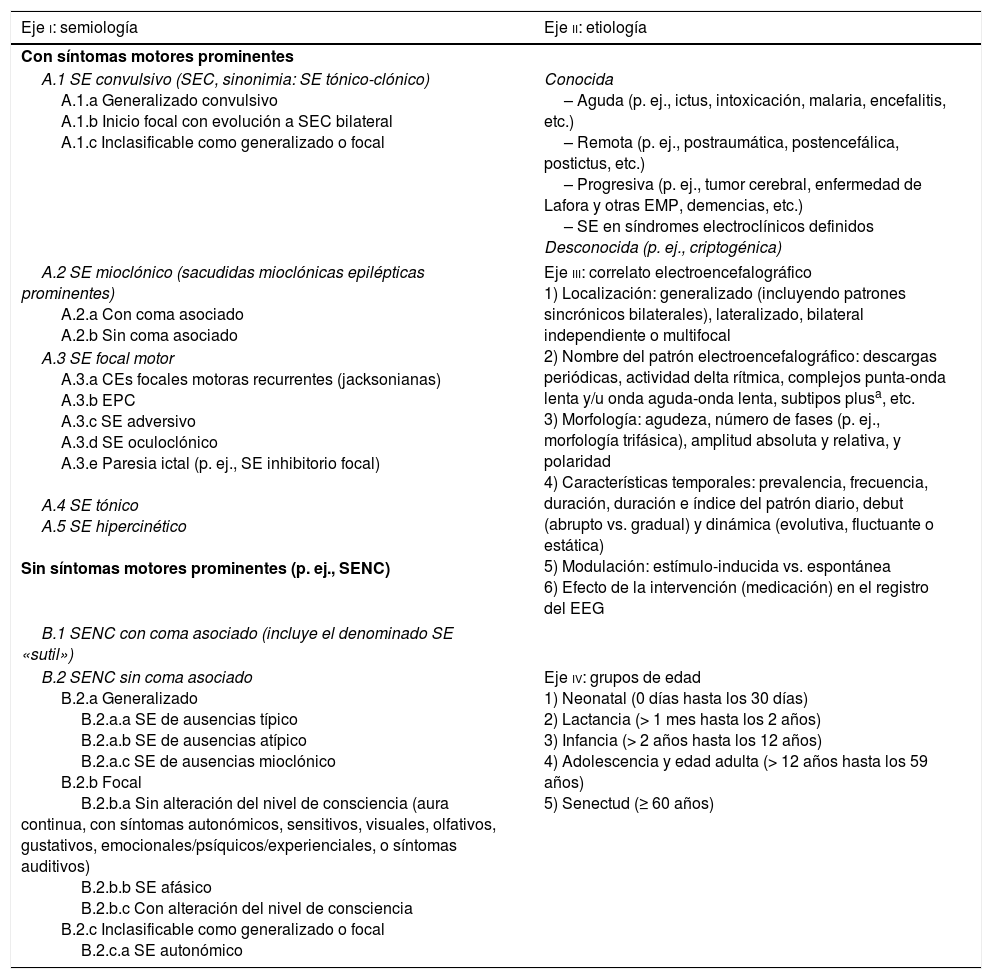
Clasificación del SE ILAE 201532–44
| Eje i: semiología | Eje ii: etiología |
|---|---|
| Con síntomas motores prominentes | |
| A.1 SE convulsivo (SEC, sinonimia: SE tónico-clónico) A.1.a Generalizado convulsivo A.1.b Inicio focal con evolución a SEC bilateral A.1.c Inclasificable como generalizado o focal | Conocida – Aguda (p. ej., ictus, intoxicación, malaria, encefalitis, etc.) – Remota (p. ej., postraumática, postencefálica, postictus, etc.) – Progresiva (p. ej., tumor cerebral, enfermedad de Lafora y otras EMP, demencias, etc.) – SE en síndromes electroclínicos definidos Desconocida (p. ej., criptogénica) |
| A.2 SE mioclónico (sacudidas mioclónicas epilépticas prominentes) A.2.a Con coma asociado A.2.b Sin coma asociado | Eje iii: correlato electroencefalográfico 1) Localización: generalizado (incluyendo patrones sincrónicos bilaterales), lateralizado, bilateral independiente o multifocal 2) Nombre del patrón electroencefalográfico: descargas periódicas, actividad delta rítmica, complejos punta-onda lenta y/u onda aguda-onda lenta, subtipos plusa, etc. 3) Morfología: agudeza, número de fases (p. ej., morfología trifásica), amplitud absoluta y relativa, y polaridad 4) Características temporales: prevalencia, frecuencia, duración, duración e índice del patrón diario, debut (abrupto vs. gradual) y dinámica (evolutiva, fluctuante o estática) 5) Modulación: estímulo-inducida vs. espontánea 6) Efecto de la intervención (medicación) en el registro del EEG |
| A.3 SE focal motor A.3.a CEs focales motoras recurrentes (jacksonianas) A.3.b EPC A.3.c SE adversivo A.3.d SE oculoclónico A.3.e Paresia ictal (p. ej., SE inhibitorio focal) A.4 SE tónico A.5 SE hipercinético Sin síntomas motores prominentes (p. ej., SENC) | |
| B.1 SENC con coma asociado (incluye el denominado SE «sutil») | |
| B.2 SENC sin coma asociado B.2.a Generalizado B.2.a.a SE de ausencias típico B.2.a.b SE de ausencias atípico B.2.a.c SE de ausencias mioclónico B.2.b Focal B.2.b.a Sin alteración del nivel de consciencia (aura continua, con síntomas autonómicos, sensitivos, visuales, olfativos, gustativos, emocionales/psíquicos/experienciales, o síntomas auditivos) B.2.b.b SE afásico B.2.b.c Con alteración del nivel de consciencia B.2.c Inclasificable como generalizado o focal B.2.c.a SE autonómico | Eje iv: grupos de edad 1) Neonatal (0 días hasta los 30 días) 2) Lactancia (> 1 mes hasta los 2 años) 3) Infancia (> 2 años hasta los 12 años) 4) Adolescencia y edad adulta (> 12 años hasta los 59 años) 5) Senectud (≥ 60 años) |
El SE es una condición resultante del fallo de los mecanismos responsables del cese de una CE y/o bien de la puesta en marcha de mecanismos que conduzcan a CEs prolongadas (después del tiempo t1). Es una condición que puede entrañar consecuencias a largo plazo (después de t2), incluyendo: muerte neuronal, daño neuronal y alteración de las redes neuronales, dependiendo del tipo y duración de las CEs. Esta definición es conceptual, con 2 dimensiones temporales: la primera es la duración de la CE y el momento temporal (t1) desde el que la CE debe considerarse como una «CE anormalmente prolongada» y, por tanto, cuando debe iniciarse el tratamiento con FAEs, prefijado en 5 min para el SE generalizado tónico-clónico, en 10 min para el SE focal (con o sin alteración del nivel de consciencia) y en 10-15 min para el SE de ausencias. La segunda es el punto temporal (t2), que es el tiempo a partir del cual se considera que la continuación de la actividad epiléptica puede comportar un riesgo de repercusiones a largo plazo, prefijado en 30 min para el SE generalizado tónico-clónico32,44.
Nota: aunque el SE generalizado convulsivo y no convulsivo con coma asociado muestran una casi perfecta correspondencia a través de la clasificación semiológica del SE, el SE focal es notoriamente heterogéneo y parece estar mejor reflejado en esta nueva clasificación de la ILAE del grupo de Trinka, ofreciendo subdivisiones clínicamente más relevantes, que también difieren en las tasas de mortalidad. Este conocimiento refinado puede permitir el desarrollo de puntuaciones clínicas pronósticas que sean más precisas que las herramientas ya existentes, y deben ser tenidas en cuenta de cara a la realización de futuros estudios epidemiológicos en este ámbito42.
CE: crisis epiléptica; EEG: electroencefalograma; EMPs: epilepsias mioclónicas progresivas; EPC: epilepsia parcial continua; FAEs: fármacos antiepilépticos; ILAE: International League Against Epilepsy; SE: status epilepticus; SENC: SE no convulsivo.
Diagnóstico1–119 Las evidencias existentes en torno al diagnóstico y tratamiento de las CEs en pacientes en tratamiento con CP son muy escasas, por lo que se deben extrapolar de la población general o de pacientes con cáncer1. El primer paso en el diagnóstico de una CE es su reconocimiento como tal, precisando su distinción de otros tipos de contracciones musculares involuntarias episódicas (p. ej., mioclono inducido por opioides), hipercinesias (p. ej., inducidas por haloperidol u ortopramidas) o trastornos del nivel de consciencia relacionados con elevación de la presión intracraneal, lo cual aparece en el 85-94% de pacientes1,102. Para ello es fundamental obtener una descripción minuciosa del episodio. Este paso es prácticamente simultáneo a la toma de decisiones terapéuticas. El diagnóstico etiológico después de una CE exige: a) anamnesis y exploración física detalladas para descartar otras entidades distractoras no epilépticas (síncope, ataque isquémico transitorio, crisis psicógena no epiléptica, amnesia global transitoria, etc.)20–22 y, en el caso de tratarse de una CE, posibles factores desencadenantes: falta de cumplimiento y/o cambios de medicación antiepiléptica (causa más importante a descartar en pacientes epilépticos conocidos), cambios del ritmo sueño-vigilia, infecciones, enfermedades sistémicas, causa farmacológica, consumo de tóxicos, estrés, luz estroboscópica, menstruación, etc. B) determinaciones analíticas (hemograma y bioquímica con glucemia, perfil hepatorrenal con Na, K, Ca, Mg, lactacto, etc.), niveles plasmáticos de medicación antiepiléptica, tóxicos en orina, gasometría arterial, EEG y pruebas de neuroimagen (tomografía computarizada [TAC] con contraste en Urgencias, aunque es de elección la resonancia magnética craneal, por su mayor sensibilidad diagnóstica). Debiendo ser conocedores de las indicaciones de: 1) TAC craneal urgente: paciente adulto no epiléptico conocido (siempre) y en el paciente epiléptico ya estudiado en caso de traumatismo craneoencefálico severo (puntuación en la Glasgow Coma Scale ≤ 8), historia de ataque isquémico transitorio o ictus, focalidad neurológica no conocida previamente, sospecha de infección del SNC, cáncer o inmunodeficiencia (p. ej., infección por el virus de la inmunodeficiencia humana [VIH]), tratamiento anticoagulante, sospecha de hemorragia subaracnoidea y/o SE sin causa obvia; 2) EEG urgente: estados confusionales prolongados, coma de origen desconocido (hasta el 8% de los pacientes en coma atendidos en Unidades de Cuidados Intensivos [UCI] y el 37% de pacientes hospitalizados, sin ninguna manifestación clínica de actividad epiléptica, corresponden a un SENC)22,41,117, retraso de recuperación del nivel de consciencia tras un SE, episodios de pérdida de consciencia breve de origen desconocido (descartar ausencias), CEs postraumáticas agudas (aumenta la probabilidad de padecer una epilepsia postraumática si se objetiva un foco irritativo en el EEG en la fase aguda [1.ª semana])118 y/o encefalitis herpética (visualizar descargas epileptiformes lateralizadas y periódicas, apoya el diagnóstico, sin embargo, también aparecen en ictus, neoplasias, encefalopatía postanóxica, etc.)119; en casos de elevada sospecha clínica y primer EEG normal, se recomienda realizar de forma secuencial otro EEG, EEG con privación de sueño, EEG de sueño y vídeo-EEG de larga duración26; y 3) punción lumbar (PL): si no hay lesiones radiológicas ni causas metabólicas, se debe realizar una PL que descarte patología infecciosa y/o carcinomatosis meníngea. La realización de las pruebas anteriormente citadas se debe individualizar en función del estado del paciente y de las preferencias de este y/o su familia.
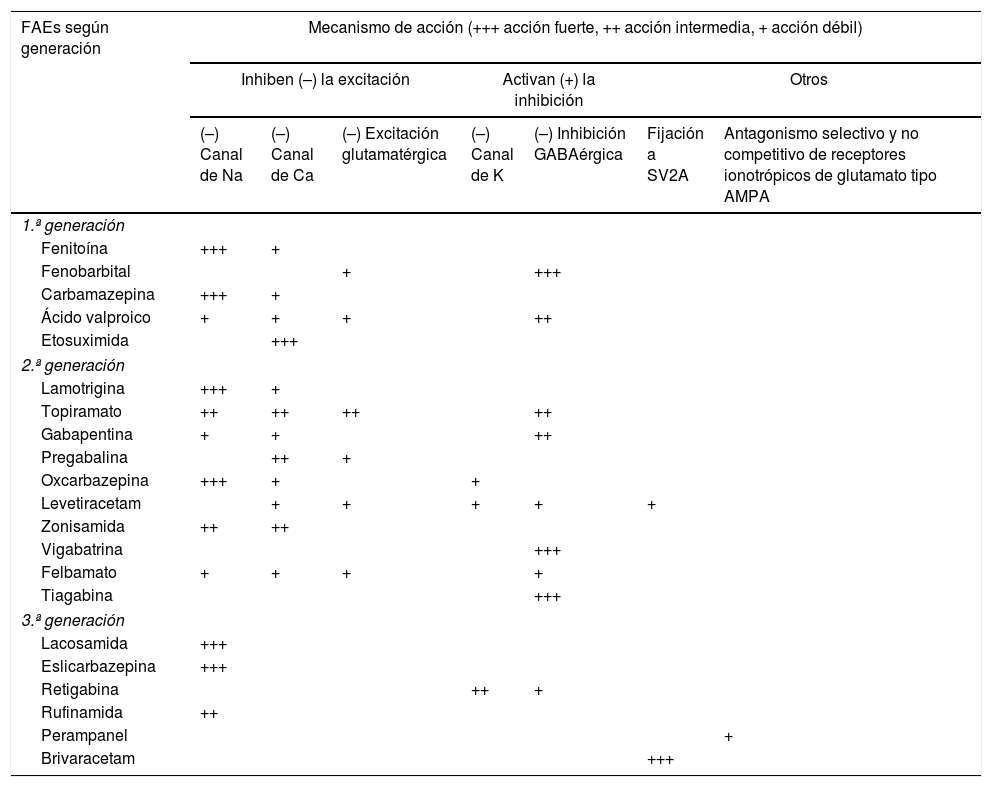
Tratamiento1–160Fármacos antiepilépticos (FAEs)27,31Los FAEs representan la base del tratamiento actual de las CEs y la epilepsia, el cual continúa siendo sintomático, sin disponer hasta la fecha actual de fármacos con actividad antiepileptogénica. En la tabla 8 se realiza una síntesis de los diversos mecanismos de acción de los FAEs para, de esta forma, entender mejor sus indicaciones y posibles asociaciones:
FAEs y mecanismo de acción27,49,90,154–158
| FAEs según generación | Mecanismo de acción (+++ acción fuerte, ++ acción intermedia, + acción débil) | ||||||
|---|---|---|---|---|---|---|---|
| Inhiben (–) la excitación | Activan (+) la inhibición | Otros | |||||
| (–) Canal de Na | (–) Canal de Ca | (–) Excitación glutamatérgica | (–) Canal de K | (–) Inhibición GABAérgica | Fijación a SV2A | Antagonismo selectivo y no competitivo de receptores ionotrópicos de glutamato tipo AMPA | |
| 1.ª generación | |||||||
| Fenitoína | +++ | + | |||||
| Fenobarbital | + | +++ | |||||
| Carbamazepina | +++ | + | |||||
| Ácido valproico | + | + | + | ++ | |||
| Etosuximida | +++ | ||||||
| 2.ª generación | |||||||
| Lamotrigina | +++ | + | |||||
| Topiramato | ++ | ++ | ++ | ++ | |||
| Gabapentina | + | + | ++ | ||||
| Pregabalina | ++ | + | |||||
| Oxcarbazepina | +++ | + | + | ||||
| Levetiracetam | + | + | + | + | + | ||
| Zonisamida | ++ | ++ | |||||
| Vigabatrina | +++ | ||||||
| Felbamato | + | + | + | + | |||
| Tiagabina | +++ | ||||||
| 3.ª generación | |||||||
| Lacosamida | +++ | ||||||
| Eslicarbazepina | +++ | ||||||
| Retigabina | ++ | + | |||||
| Rufinamida | ++ | ||||||
| Perampanel | + | ||||||
| Brivaracetam | +++ | ||||||
AMPA: ácido alfa-amino-3-hidroxi-5-metil-4-isoxazolpropiónico; Ca: calcio; FAEs: fármacos antiepilépticos; K: potasio; Na: sodio; SV2A: proteína 2A de la vesícula sináptica (involucrada en la fusión de las vesículas y la exocitosis de neurotransmisores).
Adaptada con permiso de Fernández Alonso, 201327.
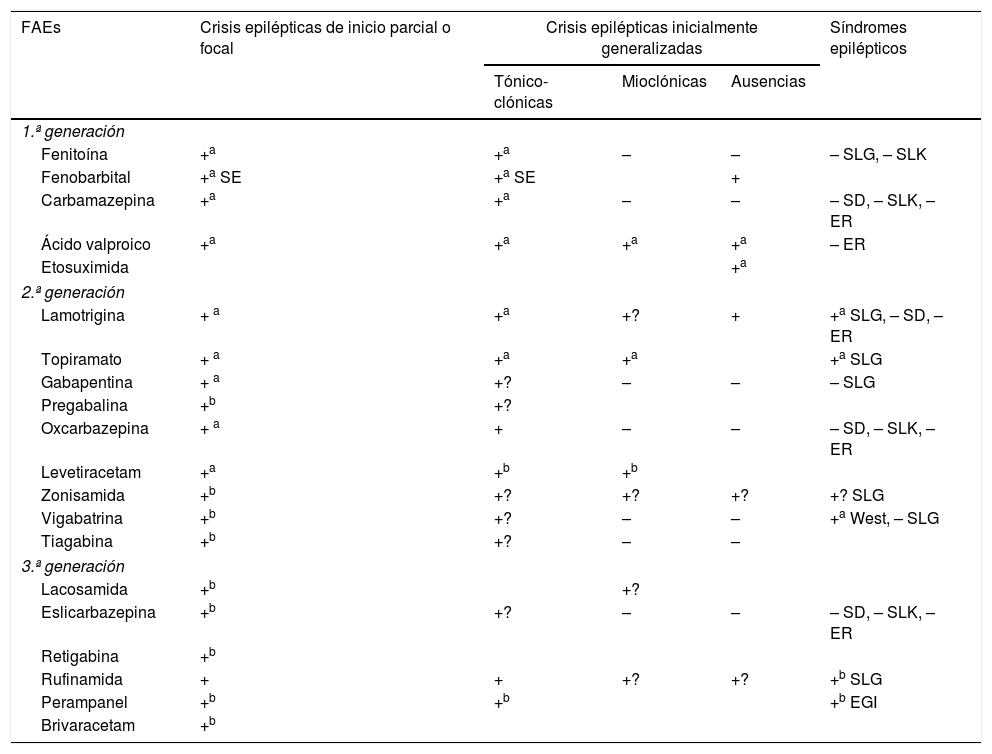
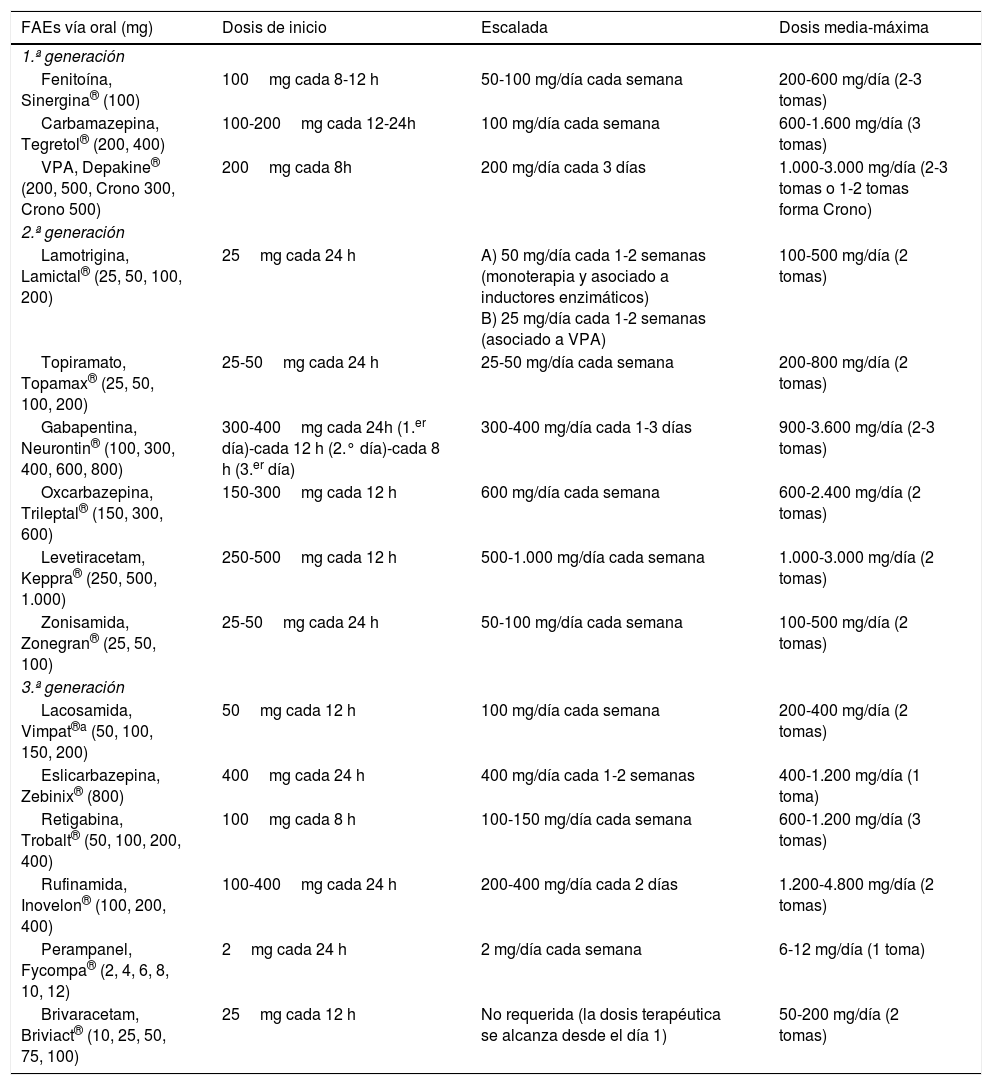
Los criterios de selección del «FAE ideal» en Urgencias son: a) buen perfil farmacológico: absorción rápida y completa por vía oral, cinética lineal, escasa unión a proteínas plasmáticas, metabolización no hepática, ausencia de metabolitos activos y de interacciones, eliminación renal y vida media prolongada. Según estas características, los FAEs con una farmacocinética más próxima a la ideal son levetiracetam (LEV), lacosamida (LCM), gabapentina (GBP) y pregabalina (PGB). Con cinética intermedia se sitúan: eslicarbazepina (ESL), lamotrigina (LTG), oxcarbazepina (OXC), retigabina (RTG), topiramato (TPM), zonisamida (ZNS) y rufinamida (RFN). Por último, los que peor cinética tienen son sobre todo la fenitoína (PHT), seguida de carbamazepina (CBZ), ácido valproico (VPA), felbamato (FBM), primidona (PRM) y tiabagina (TGB). B) Disponibilidad parenteral, cómoda conversión oral y posibilidad de terapia secuencial: necesitamos FAEs que puedan ser administrados por vía parenteral (preferentemente intravenosa [iv]), debido, entre otras razones, a la frecuente aparición de disfagia en los pacientes en SET (85%)85,89, tanto de forma directa como de forma indirecta por el deterioro del nivel de consciencia derivado de la progresión del proceso nosológico tumoral a nivel encefálico y/o por los efectos adversos del tratamiento farmacológico85,89,102,103, para alcanzar niveles terapéuticos de forma rápida tanto en situaciones de SE como de inicio de tratamiento preventivo en fase aguda. Además, en caso de ser posible, se desea poder continuar por vía oral con el mismo fármaco de forma cómoda (conversión 1:1) y segura a dosis terapéuticas. Entre los diferentes FAEs disponibles vía iv, LEV y LCM cumplen con este criterio. El VPA sería el siguiente fármaco en elegir en detrimento de PHT y los anestésicos. C) Amplio espectro de acción: lo ideal es elegir FAEs que sean capaces de controlar CEs parciales y generalizadas, debido a que con frecuencia las CEs son no presenciadas, con lo que resultan mal definidas y con anamnesis confusas. Además, no deben ser potencialmente perjudiciales para algún tipo de CE, como por ejemplo CBZ y/o PHT, que empeoran las CEs mioclónicas y las ausencias (tabla 9). Según este criterio se recomiendan LEV, LTG, VPA, TPM y/o ZNS. En pacientes con CEs de claro inicio parcial, se aconseja como primera opción LEV, LTG u OXC, y como alternativa ZNS, CBZ, TPM, GBP, LCM o ESL. En el caso de CEs generalizadas, se prefiere VPA (tónico-clónicas [GTC], mioclónicas y ausencias), LEV (CEsGTC o mioclónicas) o LTG (CEGTC o ausencias, no mioclónicas). La tabla 8 muestra el mecanismo de acción de los FAEs y la tabla 9 su eficacia según el tipo de CEs y síndromes epilépticos. D) Seguridad: criterio fundamental. Se necesitan FAEs bien tolerados, sin efectos secundarios significativos, sin interacciones con otros fármacos, incluidos otros FAEs, y que puedan ser utilizados en determinados contextos clínicos (ancianos, mujer en edad fértil, comorbilidad cardíaca, hepática, renal, etc.). En estas circunstancias, según la evidencia actual basada en series de casos, análisis retrospectivos y/u opiniones de expertos se aconseja, con excepciones, LEV, LTG, OXC, TPM, VPA y GPB en monoterapia; y LCM, perampanel y brivaracetam (BRV) como terapia coadyuvante. Se desaconseja PHT y fenobarbital (PB) de forma global, y CBZ y VPA si coexisten hepatopatía y/o polifarmacia. En insuficiencia renal, se debe tener precaución (ajustar dosis) con GBP, CBZ y derivados, TPM, LEV, BRV y LCM (tablas 10 y 11)27,45–49,59–63,74,80,89,96,100,152–159.
Eficacia e indicaciones de los FAEs27,49,97,154–158
| FAEs | Crisis epilépticas de inicio parcial o focal | Crisis epilépticas inicialmente generalizadas | Síndromes epilépticos | ||
|---|---|---|---|---|---|
| Tónico-clónicas | Mioclónicas | Ausencias | |||
| 1.ª generación | |||||
| Fenitoína | +a | +a | – | – | – SLG, – SLK |
| Fenobarbital | +a SE | +a SE | + | ||
| Carbamazepina | +a | +a | – | – | – SD, – SLK, – ER |
| Ácido valproico | +a | +a | +a | +a | – ER |
| Etosuximida | +a | ||||
| 2.ª generación | |||||
| Lamotrigina | + a | +a | +? | + | +a SLG, – SD, – ER |
| Topiramato | + a | +a | +a | +a SLG | |
| Gabapentina | + a | +? | – | – | – SLG |
| Pregabalina | +b | +? | |||
| Oxcarbazepina | + a | + | – | – | – SD, – SLK, – ER |
| Levetiracetam | +a | +b | +b | ||
| Zonisamida | +b | +? | +? | +? | +? SLG |
| Vigabatrina | +b | +? | – | – | +a West, – SLG |
| Tiagabina | +b | +? | – | – | |
| 3.ª generación | |||||
| Lacosamida | +b | +? | |||
| Eslicarbazepina | +b | +? | – | – | – SD, – SLK, – ER |
| Retigabina | +b | ||||
| Rufinamida | + | + | +? | +? | +b SLG |
| Perampanel | +b | +b | +b EGI | ||
| Brivaracetam | +b | ||||
EGI: epilepsia generalizada idiopática; ER: epilepsia rolándica; FAEs: fármacos antiepilépticos; SD: síndrome de Dravet; SE: sólo en status epilepticus; SLG: síndrome de Lennox-Gastaut; SLK: síndrome de Landau-Kleffner; +: acción eficaz; +?: eficacia dudosa; –: perjudicial.
a Indicado en monoterapia.
b Indicado como terapia coadyuvante.
Adaptada con permiso, de Fernández Alonso, 201327.
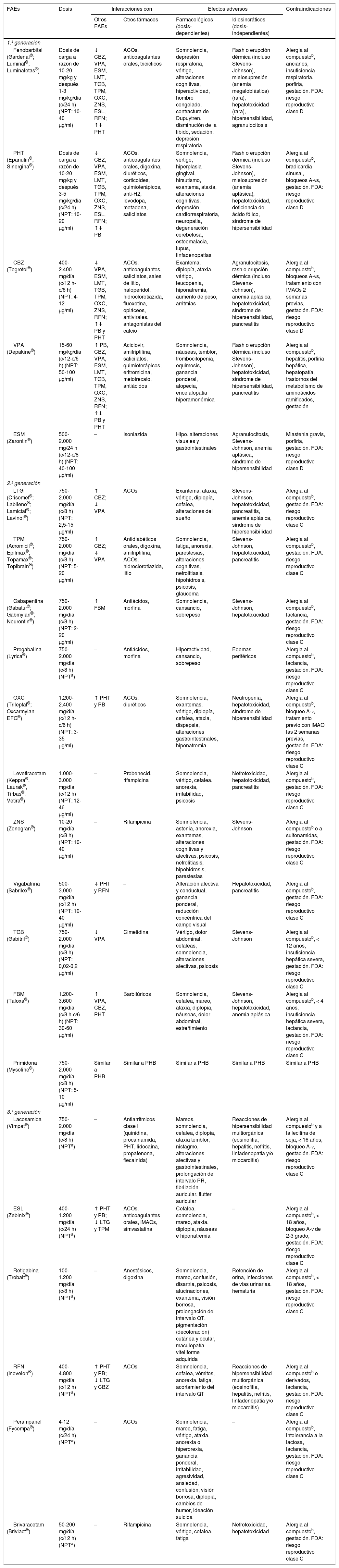
FAEs utilizados en pacientes en tratamiento con CP, especialmente de perfil oncológico (interacciones, efectos secundarios y contraindicaciones)27,45,46,49,60–64,75,81,90,97,101,154–159
| FAEs | Dosis | Interacciones con | Efectos adversos | Contraindicaciones | ||
|---|---|---|---|---|---|---|
| Otros FAEs | Otros fármacos | Farmacológicos (dosis-dependientes) | Idiosincráticos (dosis-independientes) | |||
| 1.ª generación | ||||||
| Fenobarbital (Gardenal®; Luminal®; Luminaletas®) | Dosis de carga a razón de 10-20 mg/kg y después 1-3 mg/kg/día (c/24 h) (NPT: 10-40 μg/ml) | ↓ CBZ, VPA, ESM, LMT, TGB, TPM, OXC, ZNS, ESL, RFN; ↑↓ PHT | ACOs, anticoagulantes orales, tricíclicos | Somnolencia, depresión respiratoria, vértigo, alteraciones cognitivas, hiperactividad, hombro congelado, contractura de Dupuytren, disminución de la libido, sedación, depresión respiratoria | Rash o erupción dérmica (incluso Stevens-Johnson), mielosupresión (anemia megaloblástica) (rara), hepatotoxicidad (rara), hipersensibilidad, agranulocitosis | Alergia al compuestob, ancianos, insuficiencia respiratoria, porfiria, gestación. FDA: riesgo reproductivo clase D |
| PHT (Epanutin®; Sinergina®) | Dosis de carga a razón de 10-20 mg/kg y después 3-5 mg/kg/día (c/24 h) (NPT: 10-20 μg/ml) | ↓ CBZ, VPA, ESM, LMT, TGB, TPM, OXC, ZNS, ESL, RFN; ↑↓ PB | ACOs, anticoagulantes orales, digoxina, diuréticos, corticoides, quimioterápicos, anti-H2, levodopa, metadona, salicilatos | Somnolencia, vértigo, hiperplasia gingival, hirsutismo, exantema, ataxia, alteraciones cognitivas, depresión cardiorrespiratoria, neuropatía, degeneración cerebelosa, osteomalacia, lupus, linfadenopatías | Rash o erupción dérmica (incluso Stevens-Johnson), mielosupresión (anemia aplásica), hepatotoxicidad, deficiencia de ácido fólico, síndrome de hipersensibilidad | Alergia al compuestob, bradicardia sinusal, bloqueos A-vs, gestación. FDA: riesgo reproductivo clase D |
| CBZ (Tegretol®) | 400-2.400 mg/día (c/12 h-c/6 h) (NPT: 4-12 μg/ml) | ↓ VPA, ESM, LMT, TGB, TPM, OXC, ZNS, RFN; ↑↓ PB y PHT | ACOs, anticoagulantes, salicilatos, sales de litio, haloperidol, hidroclorotiazida, fluoxetina, opiáceos, antivirales, antagonistas del calcio | Exantema, diplopía, ataxia, vértigo, leucopenia, hiponatremia, aumento de peso, arritmias | Agranulocitosis, rash o erupción dérmica (incluso Stevens-Johnson), anemia aplásica, hepatotoxicidad, síndrome de hipersensibilidad, pancreatitis | Alergia al compuestob, bloqueos A-vs, tratamiento con IMAOs 2 semanas previas, gestación. FDA: riesgo reproductivo clase D |
| VPA (Depakine®) | 15-60 mg/kg/día (c/12-c/6 h) (NPT: 50-100 μg/ml) | ↑ PB, CBZ, VPA, ESM, LMT, TGB, TPM, OXC, ZNS, RFN; ↑↓ PB y PHT | Aciclovir, amitriptilina, salicilatos, quimioterápicos, eritromicina, metotrexato, antiácidos | Somnolencia, náuseas, temblor, trombocitopenia, equimosis, ganancia ponderal, alopecia, encefalopatía hiperamonémica | Rash o erupción dérmica (incluso Stevens-Johnson), hepatotoxicidad, síndrome de hipersensibilidad, pancreatitis | Alergia al compuestob, hepatitis, porfiria hepática, hepatopatía, trastornos del metabolismo de aminoácidos ramificados, gestación |
| ESM (Zarontin®) | 500-2.000 mg/24 h (c/12-c/8 h) (NPT: 40-100 μg/ml) | – | Isoniazida | Hipo, alteraciones visuales y gastrointestinales | Agranulocitosis, Stevens-Johnson, anemia aplásica, síndrome de hipersensibilidad | Miastenia gravis, porfiria, gestación. FDA: riesgo reproductivo clase D |
| 2.ª generación | ||||||
| LTG (Crisomet®; Labileno®; Lamictal®; Lavinol®) | 750-2.000 mg/día (c/8 h) (NPT: 2,5-15 μg/ml) | ↑ CBZ; ↓ VPA | ACOs | Exantema, ataxia, vértigo, diplopía, cefalea, alteraciones del sueño | Stevens-Johnson, hepatotoxicidad, pancreatitis, anemia aplásica, síndrome de hipersensibilidad | Alergia al compuestob, gestación. FDA: riesgo reproductivo clase C |
| TPM (Acromicil®; Epilmax®; Topamax®; Topibrain®) | 750-2.000 mg/día (c/8 h) (NPT: 5-20 μg/ml) | ↑ CBZ; ↓ VPA | Antidiabéticos orales, digoxina, amitriptilina, ACOs, hidroclorotiazida, litio | Somnolencia, fatiga, anorexia, parestesias, alteraciones cognitivas, nefrolitiasis, hipohidrosis, psicosis, glaucoma | Stevens-Johnson, hepatotoxicidad, pancreatitis | Alergia al compuestob, gestación. FDA: riesgo reproductivo clase C |
| Gabapentina (Gabatur®; Gabmylan®; Neurontin®) | 750-2.000 mg/día (c/8 h) (NPT: 2-20 μg/ml) | ↑ FBM | Antiácidos, morfina | Somnolencia, cansancio, sobrepeso | Stevens-Johnson, hepatotoxicidad | Alergia al compuestob, lactancia, gestación. FDA: riesgo reproductivo clase C |
| Pregabalina (Lyrica®) | 750-2.000 mg/día (c/8 h) (NPTa) | – | Antiácidos, morfina | Hiperactividad, cansancio, sobrepeso | Edemas periféricos | Alergia al compuestob, lactancia, gestación. FDA: riesgo reproductivo clase C |
| OXC (Trileptal®; Oxcarmylan EFG®) | 1.200-2.400 mg/día (c/12 h-c/6 h) (NPT: 3-35 μg/ml) | ↑ PHT y PB | ACOs, diuréticos | Somnolencia, exantemas, vértigo, diplopía, cefalea, ataxia, dispepsia, alteraciones gastrointestinales, hiponatremia | Neutropenia, hepatotoxicidad, síndrome de hipersensibilidad | Alergia al compuestob, bloqueo A-v, tratamiento previo con IMAO las 2 semanas previas, gestación. FDA: riesgo reproductivo clase C |
| Levetiracetam (Keppra®, Laurak®, Tirbas®, Vetira®) | 1.000-3.000 mg/día (c/12 h) (NPT: 12-46 μg/ml) | – | Probenecid, rifampicina | Somnolencia, vértigo, cefalea, anorexia, irritabilidad, psicosis | Nefrotoxicidad, hepatotoxicidad, pancreatitis | Alergia al compuestob, gestación. FDA: riesgo reproductivo clase C |
| ZNS (Zonegran®) | 10-20 mg/día (c/8 h) (NPT: 10-40 μg/ml) | – | Rifampicina | Somnolencia, astenia, anorexia, exantemas, alteraciones cognitivas y afectivas, psicosis, nefrolitiasis, hipohidrosis, parestesias | Stevens-Johnson | Alergia al compuestob o a sulfonamidas, gestación. FDA: riesgo reproductivo clase C |
| Vigabatrina (Sabrilex®) | 500-3.000 mg/día (c/12 h) (NPT: 10-40 μg/ml) | ↓ PHT y RFN | – | Alteración afectiva y conductual, ganancia ponderal, reducción concéntrica del campo visual | Hepatotoxicidad, pancreatitis | Alergia al compuestob, gestación. FDA: riesgo reproductivo clase C |
| TGB (Gabitril®) | 750-2.000 mg/día (c/8 h) (NPT: 0,02-0,2 μg/ml) | ↓ VPA | Cimetidina | Vértigo, dolor abdominal, cefaleas, somnolencia, alteraciones afectivas, psicosis | Stevens-Johnson | Alergia al compuestob, < 12 años, insuficiencia hepática severa, gestación. FDA: riesgo reproductivo clase C |
| FBM (Taloxa®) | 1.200-3.600 mg/día (c/8 h-c/6 h) (NPT: 30-60 μg/ml) | ↑ VPA, CBZ, PHT | Barbitúricos | Somnolencia, cefalea, mareo, ataxia, diplopía, náuseas, dolor abdominal, estreñimiento | Stevens-Johnson, hepatotoxicidad, anemia aplásica | Alergia al compuestob, < 4 años, insuficiencia hepática severa, lactancia, gestación. FDA: riesgo reproductivo clase C |
| Primidona (Mysoline®) | 750-2.000 mg/día (c/8 h) (NPT: 5-10 μg/ml) | Similar a PHB | Similar a PHB | Similar a PHB | Similar a PHB | Similar a PHB |
| 3.ª generación | ||||||
| Lacosamida (Vimpat®) | 750-2.000 mg/día (c/8 h) (NPTa) | – | Antiarrítmicos clase I (quinidina, procainamida, PHT, lidocaína, propafenona, flecainida) | Mareos, somnolencia, cefalea, diplopía, ataxia temblor, nistagmo, alteraciones afectivas y gastrointestinales, prolongación del intervalo PR, fibrilación auricular, flutter auricular | Reacciones de hipersensibilidad multiorgánica (eosinofilia, hepatitis, nefritis, linfadenopatía y/o miocarditis) | Alergia al compuestob y a la lecitina de soja, < 16 años, bloqueo A-v, gestación. FDA: riesgo reproductivo clase C |
| ESL (Zebinix®) | 400-1.200 mg/día (c/24 h) (NPTa) | ↑ PHT y PB; ↓ LTG y TPM | ACOs, anticoagulantes orales, IMAOs, simvastatina | Cefalea, somnolencia, mareo, ataxia, diplopía, náuseas e hiponatremia | – | Alergia al compuestob, < 18 años, bloqueo A-v de 2-3 grado, gestación. FDA: riesgo reproductivo clase C |
| Retigabina (Trobalt®) | 100-1.200 mg/día (c/8 h) (NPTa) | – | Anestésicos, digoxina | Somnolencia, mareo, confusión, disartria, psicosis, alucinaciones, exantema, visión borrosa, prolongación del intervalo QT, pigmentación (decoloración) cutánea y ocular, maculopatía viteliforme adquirida | Retención de orina, infecciones de vías urinarias, hematuria | Alergia al compuestob, < 18 años, gestación. FDA: riesgo reproductivo clase C |
| RFN (Inovelon®) | 400-4.800 mg/día (c/12 h) (NPTa) | ↑ PHT y PB; ↓ LTG y CBZ | ACOs | Somnolencia, cefalea, vómitos, anorexia, fatiga, acortamiento del intervalo QT | Reacciones de hipersensibilidad multiorgánica (eosinofilia, hepatitis, nefritis, linfadenopatía y/o miocarditis) | Alergia al compuestob o derivados, lactancia, gestación. FDA: riesgo reproductivo clase C |
| Perampanel (Fycompa®) | 4-12 mg/día (c/24 h) (NPTa) | – | ACOs | Somnolencia, mareo, fatiga, vértigo, ataxia, anorexia o hiperorexia, ganancia ponderal, irritabilidad, agresividad, ansiedad, confusión, visión borrosa, diplopía, cambios de humor, ideación suicida | – | Alergia al compuestob, intolerancia a la lactosa, lactancia, gestación. FDA: riesgo reproductivo clase C |
| Brivaracetam (Briviact®) | 50-200 mg/día (c/12 h) (NPTa) | – | Rifampicina | Somnolencia, vértigo, cefalea, fatiga | Nefrotoxicidad, hepatotoxicidad | Alergia al compuestob, gestación. FDA: riesgo reproductivo clase C |
ACOs: anticonceptivos orales; A-Vs: aurículo-ventriculares; CBZ: carbamazepina; ESL: eslicarbazepina; ESM: etosuximida; FAEs: fármacos antiepilépticos; FBM: felbamato; FDA: Food and Drug Administration; IMAOs: inhibidores de la monoaminooxidasa; LTG: lamotrigina; NPT: niveles plasmáticos terapéuticos; OXC: oxcarbazepina; PB: fenobarbital; PHT: fenitoína; RFN: rufinamida; TGB: tiagabina; TPM: topiramato; VPA: ácido valproico; ZNS: zonisamida.
No establecidos.
b Compuesto: fármaco y/o excipientes contenidos en la forma de presentación.
Adaptada con permiso de Fernández Alonso, 201327.
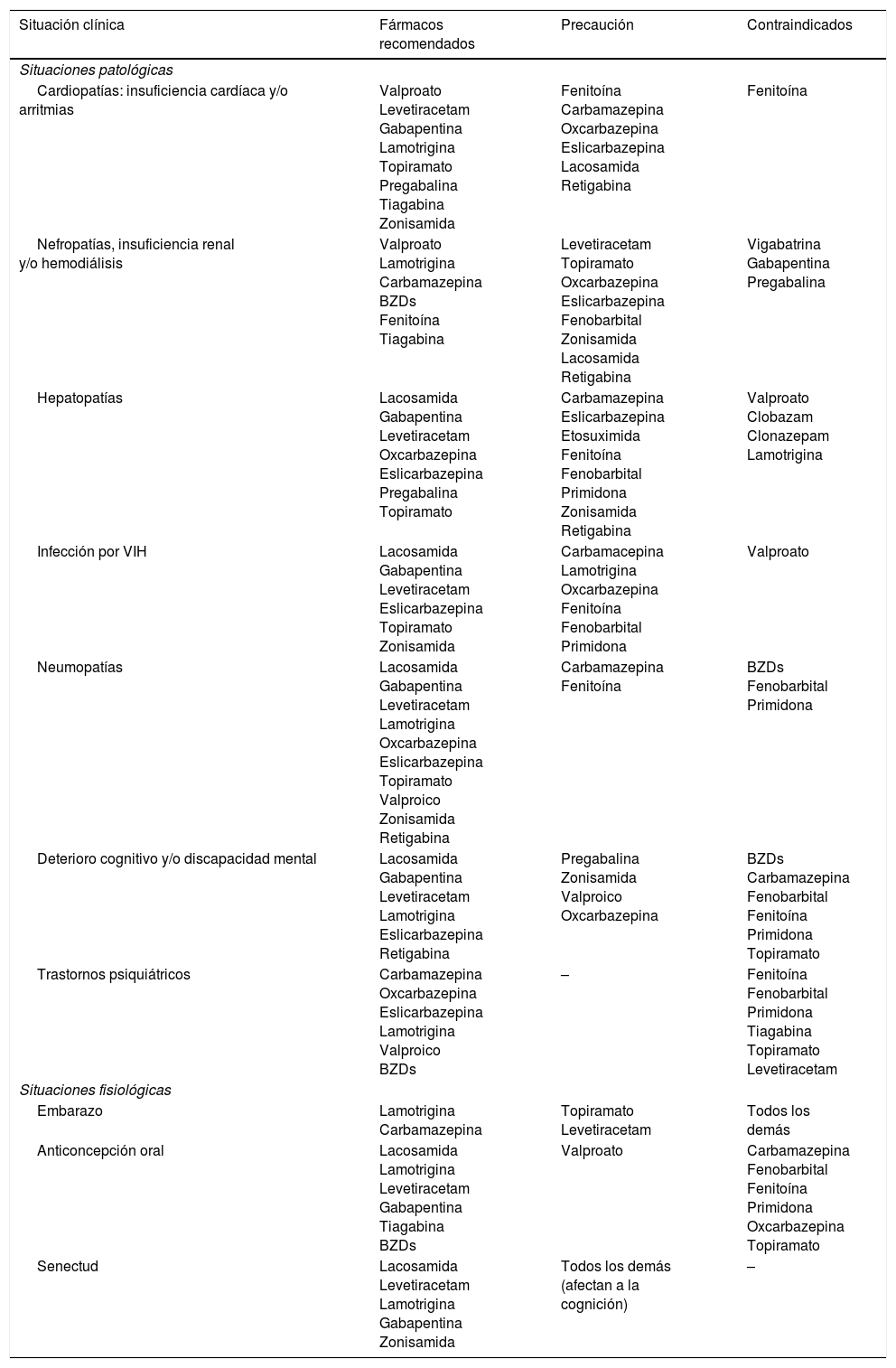
FAEs en situaciones específicas
| Situación clínica | Fármacos recomendados | Precaución | Contraindicados |
|---|---|---|---|
| Situaciones patológicas | |||
| Cardiopatías: insuficiencia cardíaca y/o arritmias | Valproato Levetiracetam Gabapentina Lamotrigina Topiramato Pregabalina Tiagabina Zonisamida | Fenitoína Carbamazepina Oxcarbazepina Eslicarbazepina Lacosamida Retigabina | Fenitoína |
| Nefropatías, insuficiencia renal y/o hemodiálisis | Valproato Lamotrigina Carbamazepina BZDs Fenitoína Tiagabina | Levetiracetam Topiramato Oxcarbazepina Eslicarbazepina Fenobarbital Zonisamida Lacosamida Retigabina | Vigabatrina Gabapentina Pregabalina |
| Hepatopatías | Lacosamida Gabapentina Levetiracetam Oxcarbazepina Eslicarbazepina Pregabalina Topiramato | Carbamazepina Eslicarbazepina Etosuximida Fenitoína Fenobarbital Primidona Zonisamida Retigabina | Valproato Clobazam Clonazepam Lamotrigina |
| Infección por VIH | Lacosamida Gabapentina Levetiracetam Eslicarbazepina Topiramato Zonisamida | Carbamacepina Lamotrigina Oxcarbazepina Fenitoína Fenobarbital Primidona | Valproato |
| Neumopatías | Lacosamida Gabapentina Levetiracetam Lamotrigina Oxcarbazepina Eslicarbazepina Topiramato Valproico Zonisamida Retigabina | Carbamazepina Fenitoína | BZDs Fenobarbital Primidona |
| Deterioro cognitivo y/o discapacidad mental | Lacosamida Gabapentina Levetiracetam Lamotrigina Eslicarbazepina Retigabina | Pregabalina Zonisamida Valproico Oxcarbazepina | BZDs Carbamazepina Fenobarbital Fenitoína Primidona Topiramato |
| Trastornos psiquiátricos | Carbamazepina Oxcarbazepina Eslicarbazepina Lamotrigina Valproico BZDs | – | Fenitoína Fenobarbital Primidona Tiagabina Topiramato Levetiracetam |
| Situaciones fisiológicas | |||
| Embarazo | Lamotrigina Carbamazepina | Topiramato Levetiracetam | Todos los demás |
| Anticoncepción oral | Lacosamida Lamotrigina Levetiracetam Gabapentina Tiagabina BZDs | Valproato | Carbamazepina Fenobarbital Fenitoína Primidona Oxcarbazepina Topiramato |
| Senectud | Lacosamida Levetiracetam Lamotrigina Gabapentina Zonisamida | Todos los demás (afectan a la cognición) | – |
Adaptada con permiso de Fernández Alonso, 201327,47,48.
BZDs: benzodiacepinas; VIH: virus de la inmunodeficiencia humana.
Aplicación de fármacos antiepilépticos (FAEs) en el ámbito de los CPs22–159 Las CEs son frecuentes en los pacientes con tumores encefálicos y su control debe ser un objetivo importante en su manejo. Los sujetos con tumores cerebrales presentan una mayor tendencia a desarrollar ER al tratamiento farmacológico. Los principales problemas que presenta el uso de FAEs en estos pacientes son los siguientes54: a) interacciones farmacológicas57,58: existen numerosas interacciones entre los FAEs y la QT, basadas en el metabolismo mediado por el CYP450. Dentro de los FAEs, existe un grupo con un potencial alto para producir interacciones (influyen y son influidos: CBZ, PHT, PB, PRM, VPA y FBM), otro grupo con un potencial medio (no influyen, pero son influidos: LTG, OXC, TGB, TPM, ESM, clonazepam [CNZ], clobazam [CLB] y ZNS) y, finalmente, un grupo con potencial bajo, que sería el ideal para emplear en CP (no influyen ni son influidos: LCM, VGB, GBP, LEV y PGB)57 (tabla 12)57–64. Por último, al inicio de la RT y/o la QT (p. ej., obleas de carmustina intraencefálicas, cisplatino intraarterial, etc.) se pueden producir CEs por irritación neuronal del área encefálica circundante90. B) Toxicidad hematológica: la neutropenia y la trombocitopenia asociadas a los FAEs clásicos son infrecuentes (0,9-1,2/104 prescripciones), pero cuando se administran junto a los citostáticos los efectos tóxicos sanguíneos se incrementan27–65,67,68,94. C) Toxicidad sobre el SNC: se debe valorar cuidadosamente el perfil de efectos secundarios de los FAEs sobre el SNC, ya que presentan una incidencia mayor que en los pacientes sin neoplasia, y la sedación, las alteraciones cognitivas, los cambios de carácter y, en algún caso, los déficits focales que pueden provocar, ya que podrían confundirse con una progresión tumoral y/o provocar un empeoramiento del estado general del paciente. D) Síndrome de hipersensibilidad: la mayor incidencia de este efecto tóxico se asocia a la administración de RT51–74. Además, estos pacientes pueden presentar estados de desnutrición, con hipoproteinemia, por lo que la fracción libre no unidad a proteínas en sangre con las dosis habituales de los FAEs clásicos, puede ser mayor de la esperada y favorecer las intoxicaciones57.
Interacciones entre fármacos antitumorales y antiepilépticos de uso más habitual55,57,58,74
| Antitumoral | Antiepiléptico | Interacción |
|---|---|---|
| IA (p. ej., aminoglutetimida) | CBZ PHT PB | ↓ [IA] |
| Capecitabina/5FU | PHT | ↑ [PHT] |
| Carboplatino | PHT | ↓ [PHT] |
| Cisplatino/doxorrubicina | CBZ, PHT | ↓ [CBZ], [PHT] |
| PHT/VPA | ↓ [VPA]/↑ [CDDP] | |
| Taxanos (p. ej., paclitaxel, docetaxel) | CBZ PB PHT | ↓ [Taxanos] |
| Inhibidores TK (–ab, –ib). | CBZ PB PHT | ↓ [Inhibidores TK] |
| Etopósido | CBZ PB | ↓ [VP16] |
| PHT/VPA | ↓ [PHT]/↑ [VP16] | |
| Tamoxifeno | CBZ PB PHT | ↓ [Tam], ↑ [PHT] |
| Alcaloides de la vinca (p. ej., vincristina) | CBZ PB PHT | ↓ [Vinca], [CBZ], [PHT] |
| Metotrexato | CBZ PHT PB | ↓ [PHT], [CBZ], [PB] ↓ [Metotrexato] |
| VPA | ↓ [VPA] | |
| Nitrosoureas (p. ej., BCNU, CCNU) | VPA | ↑ [Nitrosoureas] |
| CBZ OXC PHT PB PMD TPM | ↓ [Nitrosoureas] |
BCNU: carmustina; CBZ: carbamazepina; CCNU: lomustina; CDDP: cisplatino; IA: inhibidores de la aromatasa; PB: fenobarbital; PHT: fenitoína; PMD: primidona; TK: tirosina cinasa; Vinca: alcaloides la vinca; VPA: ácido valproico; VP16: etopósido; 5FU: 5-fluoruracilo.
Los pacientes con tumores cerebrales que reciben FAEs presentan mayor riesgo que el resto de pacientes epilépticos de presentar reacciones adversas a FAEs (RAFs). Reacciones adversas graves como el síndrome de Stevens-Johnson se asocian al uso de FAEs, especialmente durante la escalada de dosis (4-8 primeras semanas), y se han descrito con CBZ, PHT, PB, VPA, LMT, ESM, TPM, GBP, ZNS, TGB y FBM. También se han comunicado casos de Stevens-Johnson en pacientes tratados con RT craneal y que recibían simultáneamente tratamiento con PHT, CBZ y/o PB. Por lo tanto, no se recomienda el uso de estos FAEs en aquellos pacientes a los que se les esté aplicando RT holocraneal, ya que se incrementa el riesgo de presentar reacciones adversas cutáneas. Los pacientes con tumores encefálicos tratados con FAEs y sometidos a RT presentan mayor número de efectos adversos cognitivos. Se ha comprobado que estos pacientes que requieren FAEs y RT al mismo tiempo, presentan un deterioro 6 veces mayor en los tests neuropsicológicos (déficit de atención, lentitud psicomotora y/o alteración de las funciones ejecutivas), frente al grupo de pacientes que únicamente se trataron con RT, con evaluaciones a medio y largo plazo. Otros efectos adversos frecuentes son: aumento de la incidencia de cefalea con PHT, LEV y/o ZNS. Efecto mielotóxico con CBZ y/o LMT. Alteración de la esfera cognitiva y/o conductual con TPM, LEV, PHT, PB, CBZ y/o LMT. Alteración de la coordinación con PHT. Riesgo aumentado de síndrome hombro-mano en pacientes hemipléjicos tratados con PB. Como síntesis, en los pacientes con tumores cerebrales que reciban QT, RT y/o corticoides deben evitarse FAEs clásicos pos sus interacciones y/o efectos adversos idiosincráticos (nivel de evidencia [NE] iv)51.
Actitud terapéutica27,32,66–70 El tratamiento agudo de un enfermo que recibe CP, con una CE en curso presenciada, debe tratar de ser sintomático y etiológico. Se deben solucionar aquellas situaciones que entrañan riesgo vital (obstrucción de vía aérea, hipertensión intracraneal [HTIC], etc.), así como las que van a empeorar su calidad de vida (emesis incoercible, dolor refractario, etc.). Ante estos pacientes, especialmente si nos hallamos ante un enfermo en SET, han de tenerse en cuenta las siguientes premisas: a) control de los síntomas: va a ser el objetivo principal del tratamiento urgente. Cuando sea preciso y esté indicado se utilizarán mórficos y/o sedantes que mejoren el malestar del paciente y/o su familia. B) Mantener una comunicación fluida y eficaz con el paciente y/o su familia: no siempre es fácil, pero intentaremos saber qué grado de conocimiento tienen de la enfermedad que presenta el paciente al hablar con la familia, corroborar datos e informar lo más claramente posible a ambas partes, de cara a facilitar la posterior toma de decisiones conjuntamente. C) Soporte a la familia: es importante la información que suministramos y asegurarnos de que la familia comprende la situación del paciente, ya que no es infrecuente el caso de familias que se niegan a aceptar la realidad de la enfermedad. Asimismo, estos pacientes pueden necesitar en ocasiones atención a otros niveles como en el ámbito psiquiátrico, asistencia social, apoyo neuropsicológico, religioso y/o espiritual, etc.
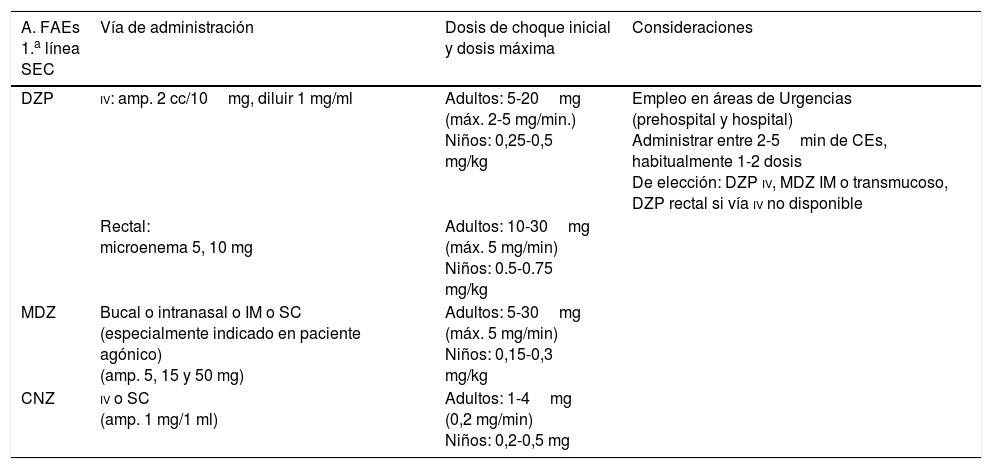
Medidas generales de soporte vital27,31,52Se toman desde el inicio de la CE, sobre todo si es convulsiva, encaminadas a estabilizar al paciente, protegerlo de posibles lesiones traumáticas derivadas, y controlar y prevenir complicaciones durante la propia CE y en el período poscrítico inmediato. Se sigue el protocolo adaptado del programa Advanced Trauma Life Support, con el esquema mnemotécnico Airway, Breathing and ventilation, Circulation, Disability, Exposure and environment (ABCDE), que consiste en: a) mantener la vía aérea permeable; b) facilitar una correcta ventilación-oxigenación; c) asegurar un control hemodinámico eficaz: monitorizar constantes vitales, canalizar una vía venosa periférica (preferiblemente dos, una para extracción analítica y otra para administración de sueroterapia y tratamiento) y corregir la causa primaria si es posible (metabólica, infecciosa), etc.; d) valorar nivel de consciencia, pupilas (una anisocoria > 1mm se considera anormal, pudiendo traducir una herniación uncal del lóbulo temporal) y función motora, y e) controlar la exposición del paciente con prevención de la hipotermia66.
Medidas para aumentar el bienestar del pacienteEn el contexto de los CP se entiende por sedación paliativa (SP), la administración de fármacos adecuados para disminuir el nivel de consciencia del paciente con el fin de reducir o anular su percepción de síntomas y/o signos que, por su elevada intensidad y/o escasa respuesta terapéutica, producen un sufrimiento innecesario (semiología refractaria [SR]). Puede ser continua o intermitente. Cuando el paciente se encuentra en SET, hablamos de sedación terminal. En esta situación, la sedación es continua. La SR que más frecuentemente justifica la SP comprende: delirium, agitación, disnea, dolor, ansiedad y hemorragia aguda o reagudizada. Los grupos farmacológicos de elección son: benzodiacepinas (BZDs) (midazolam [MDZ] y diazepam [DZP]), opioides (cloruro mórfico), neurolépticos (NLPs) sedantes (clorpromazina y levomepromazina) e incisivos (haloperidol), barbitúricos (PB) y anestésicos (propofol). Emplear un fármaco determinado se hará en función de la SR: si predominan delirium y/o agitación (1.ª elección NLPs y 2.ª elección BZDs, en especial MDZ) y si bien se trata de disnea, dolor, ansiedad y/o hemorragia (1.ª elección MDZ y 2.ª elección NLPs). Las dosis y las pautas de administración concretas escapan al objetivo de este artículo, por ello aconsejamos al lector consultar artículos, protocolos y/o GPC destinados a tal fin1,7,8,12,52,53.
Medidas para el tratamiento del edema cerebral peritumoral y la hipertensión intracraneal (HTIC)1–159 Ante una CE de comienzo reciente, el objetivo es minimizar la posibilidad de lesiones sobreañadidas. Para ello, los allegados del paciente deben ser educados acerca de cómo actuar1. El esquema básico de tratamiento de una CE en un paciente paliativo es similar al de otro paciente. La elección de un FAE debe ser individualizada, teniendo en cuenta el tipo de CE, los efectos secundarios y las posibles interacciones con otros tratamientos (QT, corticoides, etc.). El uso de corticoides puede obligar a monitorizar los niveles sanguíneos de muchos FAEs (especialmente en el caso de dexametasona [DXT] y PHT, ya que disminuyen sus niveles recíprocamente por inducción del sistema enzimático CYtochrome Pigment 450 [CYP450] hepático). En el caso de tumores (primarios y/o metastásicos) y/o radionecrosis a nivel encefálico, lo que se debe tener claro es que, ante una CE debida a estas entidades nosológicas, no está indicada la administración profiláctica de corticoides, así como tampoco en el caso de ausencia de síntomas, o signos indicativos de HTIC leve-moderada (cefalea y/o vómitos), HTIC grave (cefalea y/o vómitos intensos, alteración del nivel de consciencia y/o progresión rápida de déficits neurológicos) con déficits neurológicos estables. En el supuesto de que aparezcan síntomas y/o signos en evolución, derivados de una HTIC en pacientes oncológicos que se encuentren bajo tratamiento antiepiléptico, se instaurará una pauta de corticoides (DXT por su menor capacidad de retención salina y de inhibición de la migración leucocitaria que otros corticoides y, por tanto, con menor riesgo de sobreinfección)59, durante el menor tiempo posible a razón de 12-24 mg/24 h (inicialmente, puede administrarse un bolo de 10mg por vía iv como dosis de carga y posteriormente la dosis de 12mg —si síntomas y/o signos de HTIC leve-moderada—, 24mg —si síntomas y/o signos de HTIC grave—, distribuidas a intervalos de 4, 6 u 8 h; incrementando dicha dosis si no se obtiene mejoría transcurridas las primeras 48 h, con una reducción paulatina de 4 mg/48 h en caso de mejoría, o bien su suspensión si no se ha producido una respuesta clínica con la dosis de 24mg en 48 h) más manitol las primeras 48 h (1 mg/kg/6-8 h, manteniendo una osmolaridad plasmática de 310-320 mOsm/kg). Además de medidas posturales con elevación del cabecero de la cama > 30°, restricción de ingesta hídrica a < 1-1,5 l/día y diuréticos (furosemida 1 ampolla iv/6-8 h)52,59,65.
Medidas farmacológicas para el tratamiento de las crisis epilépticas (CEs) sintomáticas agudas27,31,52,90,91A) Los pacientes con tumores encefálicos no deben recibir profilaxis antiepiléptica si no se ha constatado una CE (grado de recomendación [GR] A)51. B) Debe iniciarse tratamiento antiepiléptico ante ≥ 2 CEs no provocadas, o ante una CE única si existe riesgo elevado de recurrencia (p. ej., CE focal y causa sintomática estructural constatada por neuroimagen y/o EEG), o bien ante un gran desasosiego del paciente y/o los familiares. C) Empezar con monoterapia a dosis bajas. D) Si la CE persiste, incrementar la dosis hasta su control o llegar a la dosis máxima tolerable. E) En pacientes con mal control de sus CEs debemos sustituir o agregar un nuevo FAE. F) Los niveles de FAEs en sangre son una guía que no debe impedir que aumentemos la dosis de FAEs prescrita si está por encima de la normalidad, pero existe un mal control de las CEs. Es importante realizar controles periódicos para valorar los niveles subóptimos. G) Elegir qué FAE usar viene determinado por el tipo de epilepsia y por los efectos adversos que comporta el uso de ese FAE. Se aconseja usar FAEs de 2.ª generación sin metabolismo hepático en pacientes con CSA por tumores cerebrales durante la RT, la QT o el tratamiento corticoideo (GR GE-SEN). H) En el caso de las mioclonías (todos los opioides pueden producirlas), se debe investigar la causa y si es tratable corregirla y/o rotar opioides. Si no se encuentra la causa y/o el enfermo está en SET, se debe instaurar tratamiento con BZDs (véase el apartado Fármacos antiepilépticos (FAEs) de primera línea).
Pauta recomendada del tratamiento con fármacos antiepilépticos (FAEs) en pacientes oncológicosNo debe administrarse de forma sistemática profilaxis antiepiléptica en los pacientes con tumores cerebrales, ya que no reduce la incidencia de CEs ni el tiempo libre de estas, e incrementa las posibilidades de presentar efectos tóxicos secundarios a los FAEs. Se acepta su uso profiláctico previo a la cirugía, pero se debe retirar progresivamente una semana después del acto quirúrgico (NE i)54,70–73,81. El perfil idóneo del fármaco a usar sería el de aquel que no interaccione con las isoenzimas del CYP450 y que se una débilmente a proteínas. A priori, estas condiciones las presentarían los nuevos FAEs, pero la experiencia en su uso es limitada y muchos de ellos no están exentos de problemas. Se propone como FAEs de primera línea el LEV (a dosis de 20 mg/kg/día, repartidos en 2 dosis) y como FAEs de segunda línea el VPA (a dosis de 15 mg/kg/día, repartidos en 3 dosis)70. Las consideraciones idiosincráticas preliminares de cada FAE, a tener en cuenta, son las siguientes27–101,120–133: la OXC es un inductor parcial. La LMT presenta frecuentemente toxicidad cutánea y el escalado de dosis es demasiado lento. El TPM puede producir alteraciones del lenguaje, parestesias y/u otros signos o síntomas focales neurológicos, además de ser inductor parcial hepático, así como provocar caquexia y cierto grado de acidosis metabólica. La GBP es un débil antiepiléptico que requiere dosis altas con riesgo de toxicidad sobre el SNC, al igual que la PGB, que, además, tiene un período de incremento de dosis lento. En el caso del VPA, fármaco inhibidor del CYP450, existe una amplia experiencia en su uso, exhibiendo una buena eficacia en el control de las CEs y una toxicidad hematológica significativamente inferior a la esperada por su mecanismo de acción. Además, la encefalopatía hiperamonémica por VPA es un efecto secundario muy infrecuente. Asimismo, se ha enunciado que a pesar del incremento de la toxicidad hematológica que acarrea su administración adyuvante con temozolamida en casos con glioblastoma, asocia un aumento de la supervivencia con respecto a quienes no toman VPA como FAE132, ya que presenta cierta actividad antineoplásica atribuida a la inhibición de la histona desacetilasa y a la reducción de la activación de la proteína cinasa C. No se recomendaría su uso cuando el paciente recibiese de forma concomitante nitrosoureas (carmustina, lomustina y se tendría que usar con precaución si se administrara junto a irinotecán. El LEV es una alternativa eficaz en las CEs secundarias a tumores encefálicos y tiene un óptimo perfil farmacocinético. Además, en estudios in vitro se ha observado que inhibe la expresión de O(6)-metilguanina-DNA metiltransferasa (enzima reparadora del ADN que desempeña un papel importante en la resistencia de las células neoplásicas a los agentes alquilantes y al citostático temozolomida)133. Asimismo, tanto VPA como LEV presentan un perfil adecuado contra la epilepsia farmacorresistente: el LEV no es sustrato del complejo proteico P-gp (glucoproteína de permeabilidad), también conocido como multidrug resistance protein 1 (MDR1), y el VPA inhibe su expresión. Ambos FAEs tienen, además, la ventaja de que se pueden administrar por vía iv. Recomendándose su asociación, en lugar de su administración secuencial en monoterapia, en pacientes con tumores cerebrales primarios y metastásicos farmacorresistentes (llegando a obtenerse un 81,5% de respondedores, una reducción de la frecuencia de CEs en el 55,6% y un 59% de pacientes libres de CEs con la combinación de VPA y LEV, manteniendo un perfil de seguridad adecuado)57,123. Otros agentes de reciente aparición como ZNS y LCM entrañan el problema de su lento ajuste de dosis y escasa experiencia en el tratamiento de la epilepsia tumoral. En cambio, la ESL y la RTG presentan un rápido ajuste de dosis, aunque sin experiencia reportada en su uso con este tipo de pacientes54.
Retirada del tratamiento antiepiléptico en pacientes oncológicos con epilepsia en remisiónEn cuanto a la controversia de mantener el tratamiento antiepiléptico en adultos con tumores encefálicos y epilepsia o plantear la retirada lenta de los FAEs, la mayoría de los autores recomienda no retirar dicho tratamiento en pacientes adultos con tumores encefálicos y que han tenido CEs, por el riesgo de recurrencia57.
Tratamiento del status epilepticus convulsivo (SEC)12,27–40,107–114 El tratamiento tiene un alto grado de éxito (80%) si se inicia dentro de los primeros 60 min. Si se retrasa más de 2 h, este disminuye al 40-50%12,34. Dependiendo del contexto clínico, puede utilizarse en primer lugar DZP iv. El DZP (0,15-0,25 mg/kg) penetra a nivel encefálico en pocos segundos, pero sus efectos antiepilépticos son breves, por lo que es preciso dar una segunda dosis 20-30 min después (máximo 20 mg). Puede administrarse por vía rectal en dosis de 5mg para niños y 10mg para adultos. Se deberá evitar la vía intramuscular (IM) por su absorción errática. Otras opciones son: lorazepam (LZP) 0,1-0,15 mg/kg iv en bolo lento (1-2 min). Puede repetirse a los 5min (esta formulación no está disponible en España). El MDZ es una alternativa y cuenta con la ventaja de su administración por vía SC. Es soluble en agua, tiene una vida media corta, el inicio de acción se produce a los 3 min tras la administración iv, 5 min tras la administración IM, 10-15 min por vía SC y 15 min por vía oral. En casos refractarios, la dosis inicial es 0,2 mg/kg, seguida por una infusión de 0,05-0,5 mg/kg/h. En pacientes ancianos, es mejor empezar con dosis de 1-2mg. En el caso de CEs en la fase de agonía, las opciones de DZP rectal y MDZ SC son especialmente útiles. Si las CEs persisten, se debe derivar al paciente, con carácter urgente, al hospital de referencia más cercano (siempre y cuando ese paciente no cumpla criterios de SET y/o haya aprobación previa del paciente y/o su familia)12,27–40 (véase el algoritmo 1 en la figura 2). El pronóstico depende principalmente del nivel consciencia al inicio, la etiología, la tipología y duración del SE, y la edad del paciente: el estupor o el coma iniciales predicen una peor recuperación neurológica, la anoxia cerebral tiene el índice de mortalidad más alto (cercano al 100%), el SEC generalizado y el SENC tienen un peor pronóstico, una duración ≥ 60 min previo a iniciar el tratamiento antiepiléptico, una edad ≥ 65 años y no tener antecedentes de CEs, comportan una mayor mortalidad110–113.
.Otras alternativas a los anestésicos: véase en el texto el apartado Fármacos antiepilépticos (FAEs) de tercera línea (coma anestésico). BIS: índice biespectral; CP: cuidados paliativos; EEG: electroencefalograma/electroencefalográfica; FAEs: fármacos antiepilépticos; IOT: intubación orotraqueal; PL: punción lumbar; SEC: status epilepticus convulsivo; VM: ventilación mecánica;. Adaptado con permiso del algoritmo de Fernández Alonso27, 2013, con modificaciones acordes con el contexto de cuidados paliativos, extraídas a partir del protocolo de la Virginia Commonwealth University")
Algoritmo 1. Manejo del status epilepticus convulsivo-SEC- (adaptado al paciente en tratamiento con cuidados paliativosespecialmente de perfil oncológico).Otras alternativas a los anestésicos: véase en el texto el apartado Fármacos antiepilépticos (FAEs) de tercera línea (coma anestésico).
BIS: índice biespectral; CP: cuidados paliativos; EEG: electroencefalograma/electroencefalográfica; FAEs: fármacos antiepilépticos; IOT: intubación orotraqueal; PL: punción lumbar; SEC: status epilepticus convulsivo; VM: ventilación mecánica;.
Adaptado con permiso del algoritmo de Fernández Alonso27, 2013, con modificaciones acordes con el contexto de cuidados paliativos, extraídas a partir del protocolo de la Virginia Commonwealth University's Thomas Palliative Care Unit, Richmond, Virginia, USA139.
- –
Benzodiacepinas (BZDs) IV: LRZ y DZP son los fármacos de elección por su mejor NE y recomendación (NE i)51 para el empleo en Urgencias. DZP tiene inicio de acción más rápido (1-3 vs. 5 min), pero su efecto dura menos tiempo (10-30min vs. 12-24 h) que LRZ, al ser este último menos liposoluble sin sufrir una rápida redistribución en los tejidos periféricos como DZP, resultando ser más eficaz44, no disponiéndose en formulación iv en España. Además de DZP, disponemos de MDZ y CNZ iv. MDZ tiene un inicio de acción más rápido (1 min) y elevada potencia, pero semivida muy corta, requiriendo perfusión continua, por lo que suele reservarse para el control del SER. Al contrario, el CNZ es más lento (3-10 min) con semivida más larga (12 h) empleándose más para terapia de mantenimiento27–40,44.
- –
Benzodiacepinas (BZDs) por vías alternativas: MDZ es el fármaco de elección para administración por vía IM, con una eficacia similar al LZP iv en el tratamiento inicial extrahospitalario (NE ii)51. En el estudio de Silbergleit et al. (aleatorizado, prehospitalario), MDZ (10mg IM) fue al menos tan eficaz como LRZ (4mg iv) en adultos, sobre todo si la vía iv no se encontraba inmediatamente accesible138; sin embargo, se debe tener en cuenta que su metabolismo por el CYP450 3A4 lo hace más proclive a presentar interacciones farmacológicas que DZP o LRZ37. En los últimos años se ha demostrado eficacia similar y cierta preferencia por la vía transmucosa (goteo oral o intranasal). Aceptado en uso pediátrico (3 meses a < 18 años) por vía transmucosa (solución bucal). MDZ no iv (bucal, nasal, IM y rectal) es igual de eficaz que DZP iv y MDZ bucal es superior a DZP rectal (NE ii)5,27–40,44,51,84,97,99,101,102,139–143: a) DZP por vía rectal: es la alternativa al MDZ no iv. Cuenta a su favor con una gran experiencia acumulada tanto en niños (canuleta de 5 mg) como en adultos (canuleta de 10 mg). B) CNZ por vía SC: es una alternativa razonable en entornos como el de los CP. C) LRZ por vía enteral (oral, o por dispositivos de sonda nasogástrica [SNG] o gastrostomía endoscópica percutánea): puede ser una buena opción en CP.
- –
Alternativas a las benzodiacepinas (BZDs): en casos de insuficiencia respiratoria y/o alto riesgo de compromiso respiratorio por sedación, o en aquellos casos en que no se recomienda la intubación orotraqueal (IOT), puede iniciarse de forma individualizada una pauta alternativa con VPA (véase dosis en SE) o lidocaína iv (bolo iv de 2 mg/kg en niños y 100-200mg en adultos) (tabla 13)27–40.
Tabla 13.A. BZDs recomendadas de primera línea en el SEC19,27–39,96,138,139. B. FAEs de segunda línea en el SEC, pauta de administración y recomendaciones prácticas19,23,27–40,136,137. C. FAEs de tercera línea en el SEC, pauta de administración y recomendaciones prácticas22,23,27–40,134,135. D. Tratamiento de los SENC22,23,27,31,40–46,102
A. FAEs
1.a línea SECVía de administración Dosis de choque inicial
y dosis máximaConsideraciones DZP iv: amp. 2 cc/10mg, diluir 1 mg/ml Adultos: 5-20mg (máx. 2-5 mg/min.)
Niños: 0,25-0,5 mg/kgEmpleo en áreas de Urgencias
(prehospital y hospital)
Administrar entre 2-5min de CEs, habitualmente 1-2 dosis
De elección: DZP iv, MDZ IM o transmucoso, DZP rectal si vía iv no disponibleRectal:
microenema 5, 10 mgAdultos: 10-30mg (máx. 5 mg/min)
Niños: 0.5-0.75 mg/kgMDZ Bucal o intranasal o IM o SC (especialmente indicado en paciente agónico)
(amp. 5, 15 y 50 mg)Adultos: 5-30mg (máx. 5 mg/min)
Niños: 0,15-0,3 mg/kgCNZ iv o SC
(amp. 1 mg/1 ml)Adultos: 1-4mg (0,2 mg/min)
Niños: 0,2-0,5 mgB. FAEs
2.a línea SECPauta de administración Recomendaciones prácticas Dosis de inicio Dosis de mantenimiento PHT
(amp. 100 y 250 mg)15-20 mg/kg a 20-50 mg/min
Por ejemplo, 1g en 250 cc de SSF 0,9% en 30min1-2 mg/kg/8 h
(tras 12 h dosis inicial)
Por ejemplo, 100 mg/8 h ivFAEs de elección en adultos
jóvenes sin comorbilidad
(cardiovascular) y establesVPA (amp. 400 mg) 15-30 mg/kg (4-6 mg/kg/min)
Por ejemplo, 1.2g con/sin diluir en 5-15min0,5-1 mg/kg/h
(tras 0,5 h dosis inicial)
Por ejemplo, 800 mg/24 hAlternativa a PHT si
contraindicada o insuficiente en pacientes sin hepatopatíaLEV
(amp. 500 mg)20 mg/kg (250-3.000 mg) en 15min
o 2-5 mg/kg/min en 100 cc SSF 0,9%
Por ejemplo, 1g en 15min o 500 mg/5min. (× 3)20-30 mg/kg/24 h
(tras 12 h dosis inicial)
máx. 3.000 mg/día
Por ejemplo, 500-1.500/12 h en 100 ccAlternativa en pacientes ancianos
y/o con comorbilidad (cardiopatía y hepatopatía) y/o refractario a PHT y/o VPALCM (amp. 200 mg) 100-400mg en 3-15min
Por ejemplo, 200mg sin diluir en 5min100-200 mg/12 h sin diluir (tras 12 h dosis inicial) FAEs opcional en pacientes refractarios a FAEs anteriores (PHT, VPA y LEV) C. FAEs
3.a línea SECPauta de administración Recomendaciones prácticas Dosis de inicio Dosis de mantenimiento Coma no barbitúrico MDZ (amp. 5, 15, 50 mg) 0,2-0,3 mg/kg en bolo (2 mg/min) 0,05-2 mg/kg/h Preferible si inestabilidad hemodinámica PPF (amp. 10, 20 mg) 1-2 mg/kg en bolo (20 μg/kg/min) 5-10 mg/kg/h (peligro si > 80 μg/kg/min) Síndrome de infusión, contraindicado en < 16 años, requiere IOT + VM Coma barbitúrico PB (amp. 200 mg) 5-20 mg/kg en 20-30min (20-50 mg/min) 2-4 mg/kg/día; 0,1-5 mg/kg/h
(a las 12-24 h dosis inicial)Alternativa al coma no barbitúrico, más eficaz, pero más efectos secundarios
Requiere IOT + VMTPT (amp. 500 mg) 2-7 mg/kg; 100-200mg en 1min (1-5 mg/kg/h) 50 mg/2-5min hasta control (0,05-2 mg/kg/h) D. SENC Tratamiento de elección Otras opciones Ausencias típicas BZD iv o mucosas (considerar opción SC en el paciente oncológico) VPA
LEV
Ausencias atípicas DZP iv
VPA/LEV iv u oralLTG
TPMParcial complejo DZP + PHT
PBCLB
LEV (de elección en el paciente oncológico)No convulsivo generalizado sutil (paciente en coma) PB MDZ (de elección en el paciente oncológico)
TPT
PPFBZDs: benzodiacepinas; CLB: clobazam; CNZ: clonazepam; DZP: diazepam; FAEs: fármacos antiepilépticos; IOT: intubación orotraqueal; LCM: lacosamida; LEV: levetiracetam; LTG: lamotrigina; MDZ: midazolam; PB: fenobarbital; PHT: fenitoína; PPF: propofol; TPM: topiramato; TPT: tiopental; VM: ventilación mecánica. VPA: ácido valproico.
A, B, C: adaptada con permiso de Fernández Alonso, 201327.
D: adaptada con permiso de Mercadé-Cerdá et al., 201323.
Las recomendaciones más recientes establecen la necesidad de administrar FAEs de forma más precoz. Siguiendo esta línea, nacen las últimas tendencias conceptuales. El retraso en el inicio de FAEs iv y/o a dosis infraterapéuticas asocia mayor refractariedad y peor pronóstico. Si la CE no se yugula con las medidas anteriores en los tiempos prefijados, estamos ante un SE, y como tal, debemos iniciar tratamiento con FAEs iv (PHT, VPA, LEV y/o LCM), sin existir estudios con NE i demostrando superioridad de uno u otro, debiendo escogerse por exclusión según comorbilidades y tolerabilidad del paciente y/o posibles interacciones farmacológicas; siendo en el paciente oncológico los FAEs iv de elección LEV, VPA y/o LCM44. Disponemos de los siguientes FAEs en segunda línea (tabla 13)27–40,44:
- –
PHT27–40,94–97: es el FAE de referencia para el SEC en segunda línea en la mayoría de guías clínicas, gracias a su dilatada experiencia y recomendación de la Food and Drug Administration (FDA). Su eficacia es mayor en SEs parciales que en generalizados, desaconsejándose en SEs mioclónicos y de ausencias. No deprime la función respiratoria ni afecta al nivel de consciencia. Su mayor limitación es su toxicidad cardiovascular (hipotensión arterial, arritmias, etc.), por lo que ha de utilizarse con gran precaución en pacientes ancianos y/o cardiópatas. También produce flebitis por su pH alcalino de 1234, debiéndose administrar por una vía diferente de las BZDs y en sueros no glucosados. Otro inconveniente es que, a máxima velocidad, la dosis necesaria tarda en administrarse unos 30min, demasiado tiempo para comprobar su efectividad y para dar un segundo FAE en caso de ineficacia, previo a plantear administrar anestésicos como tercera línea. En el paciente oncológico, dadas sus potenciales interacciones con fármacos antineoplásicos, así como su posible efecto deletéreo sobre la función linfocitaria94,95, en la actualidad se prefiere utilizar LEV y, en caso de ineficacia, asociar VPA y/o LCM27–40.
- –
VPA: a pesar de carecer de un NE i (siendo 2B), su uso está aprobado en numerosos países, basado en la experiencia acumulada de > 300 estudios publicados34, ya que ha resultado ser tan eficaz como PHT. Por ello, la mayoría de las guías lo sitúan como el FAE a añadir ante el fracaso y/o contraindicación de PHT en el SEC. En un estudio aleatorizado no ciego, con 68 pacientes en SEC, a dosis altas (30 mg/kg durante 15 min), fue más eficaz que PHT (18 mg/kg a 50 mg/min). En los pacientes refractarios al primer FAE, VPA demostró ser más eficaz (79%) que PHT (25%)144. Otorga una serie de ventajas frente a esta: mayor rapidez de acción, mejor tolerancia, sedación ligera, ausencia de cardiotoxicidad y amplio espectro de acción. No requiere ajuste en insuficiencia renal y por su alta unión a proteínas plasmáticas su metabolismo no se afecta por la diálisis. Los efectos adversos más comunes son hipotensión, mareo y/o trombocitopenia. En la práctica clínica, ha ido consolidándose como alternativa a la PHT en pacientes ancianos y/o cardiópatas. Su principal limitación es su metabolismo hepático. Está contraindicado en pacientes con enfermedades mitocondriales, hepatopatías y/o coagulopatías, porfiria, inmunosupresión y/o infección por VIH. Se desaconseja en mujeres en edad fértil por inducir síndrome de ovario poliquístico y/o efectos teratogénicos27–40.
- –
LEV: es uno de los FAEs más modernos del arsenal alternativo a PHT y VPA cuando estos estén contraindicados y/o sean ineficaces. Está disponible por vía iv desde 2007, por lo que su experiencia es más limitada. Su evidencia está basada principalmente en series de casos tratados con LEV como primer FAE de segunda línea o tras PHT y/o VPA, tanto en SEC como SENC. Las dosis efectivas son 500-3.000 mg/día, logrando el control de las CEs en 12-96 h. Se toleran hasta 1.500mg en 5min. Una pauta eficaz y segura es administrar 500mg cada 5min durante 15min. Frente a PHT y VPA, no existen estudios adecuadamente diseñados. En el trabajo de 2011 de Alvarez et al., tras un análisis ajustado a la severidad y etiología del SE, la eficacia de la PHT fue mayor que la de LEV y menor que la de VPA145. En diferentes estudios se ha reportado una eficacia similar de LEV frente a VPA en SEC y SENC de ausencias a los 30 min146–148. Sus principales ventajas son su seguridad, sin efectos adversos graves, pocas interacciones, cinética lineal y fácil administración. Por su metabolismo requiere ajuste (reducir a la mitad) en pacientes con aclaramiento de creatinina inferior a 30 ml/min. Es una buena opción como primer FAE en pacientes ancianos, oncológicos, polimedicados, con comorbilidad cardiovascular y/o hepática y una buena alternativa tras fracaso de PHT y/o VPA27. Adicionalmente, se ha invocado un efecto antiemético adscrito al uso de LEV, si bien el mecanismo de acción exacto permanece inaclarado125. Por último, en una revisión de la literatura de 2014, además de en un caso reciente, se ha reportado un balance eficacia/seguridad favorable en su uso SC como vía alternativa en CP149,150.
- –
LCM: es el FAE empleado en SE más moderno hasta el momento, disponible en España desde 2011 e inicialmente desarrollado como formulación iv. Es una nueva opción terapéutica en pacientes con SER a los fármacos anteriores, sobre todo en SEC de inicio parcial, aunque también ha dado buenos resultados en SEC generalizados. Su evidencia y recomendación son aún limitadas (3D). Se han publicado 19 estudios (10 casos clínicos y 9 series de casos) con 136 pacientes (50% en SEC, 31% SEC focal, 19% SEC generalizado) de edad entre los 8 y los 90 años, tratados con LCM refractarios a varios FAEs con éxito cercano al 60% de los pacientes, sobre todo asociado a VPA y/o LEV. Cuanto más precoz se administre, la eficacia parece mayor. Las dosis habitualmente usadas oscilaron entre 100-400mg en 3-15min. La dosis de 200-300mg en 15min mostró una eficacia similar a la de 400mg, pero más segura. A su vez, la dosis de 400mg se mostró más segura que la de 600mg (no indicada en ficha técnica) y con menos efectos secundarios. LCM mostró un buen perfil de seguridad sin efectos secundarios graves, salvo en un caso donde se detectó bloqueo A-v completo asociado a antiarrítmicos del grupo i (bloqueantes de canales de sodio [Na]). Los efectos secundarios habituales son neurológicos leves y dependientes de la dosis. Al igual que LEV, es un FAE cercano al considerado ideal en Urgencias, con un potencial óptimo en SE27–40.
- –
PB: los barbitúricos fueron los primeros fármacos utilizados en el SE, desde principios del siglo xx y hasta la llegada de las BZDs en la década de los 60, eran FAEs de primera línea. En países en vías de desarrollo, el PB continúa siendo el FAE más utilizado tras las BZDs, destacando una eficacia similar a PHT. En España está indicado en SE como alternativa a PHT. En CEs neonatales sigue siendo una buena opción. Su principal limitación es su bajo perfil de seguridad, con riesgo de inestabilidad hemodinámica y necesidad de ventilación mecánica, además de alteraciones cognitivo-conductuales. Con la aparición de nuevos FAEs iv, ha caído en desuso como FAE de segunda línea, reservándose para tercera línea como coma inducido (tabla 12)27–40.
Según las evidencias científicas en el tratamiento del SEC establecido: DZP + PHT, PB y LZP iv son igual de eficaces en el control del SEC que los 20min de inicio de la perfusión y durante la primera hora1 (NE i)51. PHT y VPA, VPA y LEV iv son igual de eficaces en el control del SEC que los 30min del comienzo de la perfusión y en efectos adversos (NE ii)51. LCM iv ha demostrado su eficacia en distintos estudios no prospectivos ni controlados y en series de casos para distintos tipos de SEC (NE iv)51. La mayoría de las GPC recomienda usar LZP (4 mg/iv) ó DZP (10 mg/iv) seguido de PHT (18 mg/kg/iv) o PB (20 mg/kg/iv) (NE iv)51. El empleo de VPA, LEV o LCM estaría indicado en el caso de contraindicación de la PHT, como alternativa al PB iv o si nos encontramos ante un SER (NE iii)51. LEV y LCM no tienen indicación autorizada para su empleo en los SE51. En cuanto a los GR, el tratamiento farmacológico inicial de cualquier CE prolongada y del SE debe realizarse con BZDs (GR A). PHT y/o PB iv deben emplearse si no hay control del SE con BZDs (GR A). VPA y/o LEV iv deben utilizarse en los SE si están contraindicados PHT y VPA (GR B). LEV y LCM pueden emplearse en los SE en lo que esté contraindicada la PHT y/o como alternativa al PB iv y/o en los SER (GR C). Ciñéndonos al paciente oncológico en CP, dentro de esta segunda línea de tratamiento, sin sobrepasar en exceso los 30min de SEC, podemos emplear por orden de elección LEV, LEV + VPA, LEV + LCM, VPA + LCM o LEV + VPA + LCM27–40. PMP se ha probado en varias series de casos pequeñas (la mayor serie reclutó a 12 pacientes) por SNG, demasiado heterogéneas para extraer conclusiones fehacientes en cuanto a su eficacia en SEC44. BRV se encuentra disponible en formato iv, y aunque los expertos en epilepsia le atribuyen un potencial inherente como FAE coadyuvante en SE, aún no existen estudios publicados al respecto en seres humanos44.
Fármacos antiepilépticos (FAEs) de tercera línea (coma anestésico)27–40,139,140En los casos en los que el SE es refractario a FAEs de segunda línea tras 30min de actividad epiléptica y tratamiento, debe plantearse la necesidad de anestésicos (siempre y cuando ese paciente no cumpla criterios de SET y/o haya aprobación previa del paciente y/o su familia). Sus efectos sistémicos (hipotensión arterial, depresión miocárdica y/o hepatotoxicidad) limitan su uso. Se recomienda su administración en la UCI, bajo monitorización con ECG-EEG y medidas de soporte vital. En caso de requerir IOT, se prefiere un relajante muscular de semivida corta, como vecuronio, para que no dificulte la valoración neurológica posterior. La elección de los fármacos inductores del coma anestésico en los SER debe basarse en la experiencia y/o los protocolos de la UCI correspondiente. En el caso del paciente oncológico en CP, dicho ingreso en UCI debe ser preferiblemente evitado102. Existen 2 opciones, coma barbitúrico o no barbitúrico. No existen estudios adecuados de superioridad de uno frente a otro (NE iv)27–40,44,51:
- –
Coma no barbitúrico (MDZ/propofol): se prefiere frente a los barbitúricos en los pacientes inestables hemodinámicamente, no por ser más eficaz, sino por su mayor seguridad y rapidez de acción. Se puede optar MDZ o propofol. Se prefiere el uso de MDZ por su mejor perfil de seguridad, si bien debe conocerse que la aparición de sus efectos adversos y la taquifilaxia son más frecuentes si se administra en infusión continua39,139. Por otro lado, aunque propofol a dosis bajas es seguro, existe riesgo de síndrome de infusión por propofol (acidosis metabólica severa, rabdomiólisis, fracaso renal, hipotensión arterial, apneas, bradicardia, etc.) potencialmente letal. Por ello, requiere monitorización de constantes vitales y está contraindicado en niños27–40,139–143.
- –
Coma barbitúrico (PB/tiopental): se suele reservar como rescate al coma no barbitúrico, evitándose en pacientes inestables hemodinámicamente por sus efectos secundarios, sobre todo hipotensión y sedación. La eficacia es similar a la conseguida con PHT. Se puede elegir PB o tiopental (produciendo su metabolito activo pentobarbital) sin claras diferencias, salvo la toxicidad cardiovascular de tiopental151 (tabla 13).
- –
Alternativas a los anestésicos: anestésicos inhalados (isoflurano, desflurano); lidocaína iv; ketamina iv; sulfato de magnesio iv (sobre todo en mujeres con eclampsia); FAEs (TPM por vía enteral; CBZ y CLB por vía oral), terapias inmunológicas (corticoides y/o inmunosupresores) y terapias no farmacológicas (dieta cetogénica, hipotermia y/o estimulador del nervio vago), entre otras27–40,44,47,81,83,89.
En la actualidad, no existe ninguna guía o consenso de tratamiento para el SENC, debido a la falta de estudios y datos de eficacia. Se utilizan las mismas recomendaciones indicadas para el SEC, pero con un NE bajo (GE-SEN)51. No se recomienda actitud agresiva en pacientes sin coma profundo, debido a que sus consecuencias no son tan dramáticas. Por tanto, se debe iniciar con BZDs y observar la respuesta. Si persiste la actividad epiléptica, se debe recurrir a los FAEs de segunda línea: son preferibles VPA y LEV en el SE de ausencias, y LEV o PHT en el SE parcial complejo. LCM parece una buena alternativa en casos refractarios a los previos por su eficacia, seguridad y amplio espectro de acción (tabla 13)27,40,41.
Resumen de manejo del status epilepticus convulsivo (SEC) (fig. 2)1–151Tratamiento preventivo antiepiléptico tras la primera CEs27- –
FAEs de inicio como tratamiento preventivo: en primer lugar, debe decidirse si es preciso iniciar tratamiento preventivo de una nueva CE (prevención secundaria) en la fase aguda a corto plazo para evitar recidivas precoces, o bien con intención de mantenerlo a largo plazo, aspecto este más difícil de determinar en el momento agudo. La estrategia de iniciar tratamiento con FAEs debe cumplir la premisa de aportar más beneficios que riesgos y debe ser consensuada con el paciente y/o su familia teniendo en cuenta sus preferencias.
- –
Riesgos y beneficios relacionados con el FAE de inicio: el comienzo de tratamiento con FAEs pretende minimizar el riesgo de recurrencias debido a que estas se asocian a mayor probabilidad de accidentes, restricciones laborales, estigma social e incluso muerte súbita, además de visitas repetidas a Urgencias. El riesgo de recurrencia es mayor tras presentar un SE o una segunda CE espontánea (70% en el primer año). Tras una sola primera CE, el riesgo es menor (40% a los 2 años). El inicio del tratamiento con FAEs tras una primera CE reduce el riesgo de recurrencia en un 34% de media, sobre todo a corto plazo (< 2 años). En cambio, no se han encontrado claras diferencias a largo plazo (> 5 años) ni tampoco mejoran claramente el pronóstico vital (NE i)27,51. Se ha descrito una serie de factores de riesgo de recurrencia: tipo de CE (inicio parcial, etiología sintomática), número de CEs (≥ 2), alteraciones en la exploración neurológica, anomalías epileptiformes en el EEG y estructurales en neuroimagen. En este sentido, se ha desarrollado un índice pronóstico de recurrencia según el estudio Multicentre trial for early Epilepsy and Single Seizures (MESS): a) 1 punto si antes de la consulta actual existieron 2 ó 3 CEs; 2 puntos si ≥ 4 CEs, y b) sumar 1 punto si existe alteración neurológica (alteraciones o déficits neurológicos, trastorno del aprendizaje o retraso en el desarrollo) y sumar otro punto si EEG anormal (alteraciones epileptiformes u ondas lentas). Según la puntuación final el riesgo será bajo (0 puntos), medio (1 punto) ó alto (2-4 puntos)152.
- –
Indicaciones de tratamiento preventivo (tabla 14)27: en general, se sugiere iniciar tratamiento tras SE, ≥ 2 CEs no provocadas o una sola crisis (provocada o no) acompañada de alteraciones en el EEG, alguna patología neurológica potencialmente epileptogénica (alteración estructural aguda y/o enfermedad previa) o gran exigencia psicosocial en el paciente. Según el índice MESS, se debe iniciar tratamiento con FAEs si el riesgo es medio-alto (≥ 1 punto). En cualquier caso, estos pacientes deben ser remitidos a una consulta de Neurología (Unidades de Epilepsia) con el objetivo de completar el estudio etiológico y de iniciar o mantener el tratamiento antiepiléptico de forma crónica.
Tabla 14.A. Resumen las principales indicaciones de FAEs en prevención secundaria. B. FAEs como tratamiento preventivo
A. Indicaciones de iniciar prevención secundaria de CEs Duración prevención secundaria Segunda CE no provocada o SE A largo plazo Primera CE si alto riesgo de recidiva CEs sintomática aguda si:
– Alteración estructural aguda (infección del SNC, ictus, TCE severo*)
– Abstinencia enólica
– EclampsiaA corto plazo (inicio en fase aguda)
* Prevención primaria (1 semana)CE sintomática remota: todas
CE de etiología indeterminada
– Inicio diferente a GTC
– Déficit neurológico
– Edades extremas de la vida
– Focalidad neurológica tras CE (p. ej., parálisis de Todd)
– Lesión en neuroimagen
– Patrón epileptiforme en EEG
A largo plazo (valorar inicio en fase aguda, previo al alta de urgencias o esperar a consulta especializada con Neurología)B. FAEs como tratamiento preventivo Consideraciones En fase aguda se recomienda (por orden de elección)
1.° LEV
2.° VPA
3.° PHT
4.° CBZ (oral)
En fase aguda se recomienda administración ivTras la fase aguda, valorar prevención secundaria con
LEV
Como alternativas
LTG
VPA
OXC
Al alta de urgencias, tras fase aguda, se recomienda continuar (si es posible), por vía oralCBZ: carbamazepina; FAEs: fármacos antiepilépticos; GTC: generalizada tónico-clónica; LEV: levetiracetam; LTG: lamotrigina; OXC: oxcarbazepina; PHT: fenitoína; SE: status epilepticus; SNC: sistema nervioso central; TCE: traumatismo craneoencefálico; VPA: ácido valproico.
Adaptada con permiso de Fernández Alonso, 201327.
- –
Elección de FAEs para iniciar tratamiento preventivo(tabla 14)27: la elección depende del tipo de CE (clasificación), del paciente (edad, comorbilidad y tratamiento habitual) y del propio FAE (eficacia, seguridad, disponibilidad y comodidad):
El paciente epiléptico descompensado puede presentarse en varios escenarios clínicos, primando en todos ellos la seguridad. Los criterios de derivación al hospital son los considerados como factores de riesgo: sospecha de nueva lesión encefálica (CEs diferentes de las habituales, exploración neurológica anormal y/o enfermedad basal como cáncer, sida, etc.), CEs prolongadas o recurrentes (SE o CEs en salvas [cluster] ≥ 3 CEs en 24 h) y/o sospecha de complicaciones sistémicas y/o traumáticas. Desde un punto de vista pragmático, se distinguen descompensaciones relacionadas con la recurrencia de CEs (eficacia) o con RAFs habituales (seguridad):
- –
Crisis epilépticas recurrentes: a) CEs similares a la habituales: pacientes sin modificación en características ni frecuencia de las CEs. La actitud en estos pacientes es conservadora, con seguimiento habitual (sin necesidad de pruebas complementarias), manteniendo o reiniciando el tratamiento habitual, poniendo especial hincapié en evitar desencadenantes y asegurar el cumplimiento terapéutico. B) Aumento de frecuencia de CEs: debemos preguntarnos si el paciente toma adecuadamente sus FAEs y si estos se encuentran en rango terapéutico. Se pueden solicitar niveles de PHT (10-20 μg/ml), PB (10-40 μg/ml), CBZ (4-12 μg/ml) y VPA (50-100 μg/ml). Si el desencadenante es claramente el abandono y/o incumplimiento terapéutico y el paciente se encontraba bien controlado con sus FAEs habituales, se recomienda retomar su tratamiento. En los casos en los que se sospecha buen cumplimiento a dosis adecuadas y los niveles están por debajo del rango, hemos de sospechar interacciones farmacológicas y/o enfermedad intercurrente. En estos casos tenemos la opción de aumentar la dosis del FAE o valorar cambio a otro sin interacciones. En cambio, si los niveles están en rango podemos aumentar la dosis y/o añadir otro FAEs nuevo. Si no disponemos de niveles y la dosis habitual es la máxima, podemos optar por añadir un nuevo FAE o esperar a la próxima consulta de Neurología.
- –
Reacciones adversas a FAEs (RAFs)27,45: a) RAFs farmacológicas (dosis-dependientes): la mayoría son efectos sobre el SNC: somnolencia, ataxia, disartria, diplopía, visión borrosa, etc. Se solucionan reduciendo las dosis y titulando más lentamente. La monitorización de niveles es útil si son niveles supraterapéuticos, esperando paralelismo entre mejoría clínica y normalización de niveles. B) RAFs idiosincrásicas (dosis-independientes): los efectos leves son relativamente frecuentes, sobre todo en forma de rash cutáneo, pero pueden producirse reacciones graves que afectan a la piel (síndrome de Stevens-Johnson), médula ósea (agranulocitosis) y/o hígado (hepatitis tóxica). La reacción de hipersensibilidad (fiebre, exantema, adenopatías, edemas, etc.) puede ser muy grave y aparece más frecuentemente con CBZ, PHT, PB, LTG, OXC y/o ZNS. Ante una reacción exantemática, conviene cambiar por uno de menor riesgo, como LEV, GBP y/o TPM. En ancianos son frecuentes los efectos cognitivos, sobre todo debido a FAEs clásicos. En mujeres en edad fértil, el VPA está contraindicado por efectos sobre su aparato reproductor y teratogénico. La hiponatremia es un problema frecuente asociado a CBZ y derivados (OXC y ESL) que ocasiona retirada y/o cambio de FAE.

Algoritmo 2. Ajuste de FAEs en el paciente epiléptico descompensado. FAE: fármaco antiepiléptico; FAEs: fármacos antiepilépticos.
Adaptado con permiso de Fernández Alonso27.
En lo referente a la politerapia racional en ER (frecuente en los pacientes oncológicos), la mayoría de autores aconseja probar los nuevos FAEs, siempre que no consigamos el control de la ER, y estén indicados. En esta situación de refractariedad, LCM es una buena opción por su mecanismo de acción novedoso y su perfil clínico, próximo al FAE ideal. La evidencia actual es limitada debido a que procede de estudios abiertos no aleatorizados, series de casos, análisis post hoc y opiniones de expertos. Según esta, se consideran combinaciones potencialmente útiles las siguientes: FAE no bloqueante de sodio (p. ej., VPA o LEV) + LCM; LCM o VPA + LEV, TPM, ZNS o LCM; CBZ, OXC, ESL o PHT + LEV, LCM, ZNS o RTG y VPA + ESM. Por el contrario, se deben vigilar CBZ, OXC, ESL o PHT + LTG; VPA + PHT y CBZ o PHT + TPM o TGB. No se recomiendan CBZ + PHT; OXC + ESL ni PRM, TGB o VGB. Por último, en cuanto a FAEs genéricos (EFG), no se recomienda intercambiar FAEs de marca por EFG en pacientes con buen control libre de CEs o epilepsias de difícil control27,153. En la tabla 15 figuran los FAEs de marca más usualmente empleados por vía oral.
FAEs de marca más frecuentemente administrados por vía oral (si es posible esta vía de administración) y su posología27,45,46,154–158
| FAEs vía oral (mg) | Dosis de inicio | Escalada | Dosis media-máxima |
|---|---|---|---|
| 1.ª generación | |||
| Fenitoína, Sinergina® (100) | 100mg cada 8-12 h | 50-100 mg/día cada semana | 200-600 mg/día (2-3 tomas) |
| Carbamazepina, Tegretol® (200, 400) | 100-200mg cada 12-24h | 100 mg/día cada semana | 600-1.600 mg/día (3 tomas) |
| VPA, Depakine® (200, 500, Crono 300, Crono 500) | 200mg cada 8h | 200 mg/día cada 3 días | 1.000-3.000 mg/día (2-3 tomas o 1-2 tomas forma Crono) |
| 2.ª generación | |||
| Lamotrigina, Lamictal® (25, 50, 100, 200) | 25mg cada 24 h | A) 50 mg/día cada 1-2 semanas (monoterapia y asociado a inductores enzimáticos) B) 25 mg/día cada 1-2 semanas (asociado a VPA) | 100-500 mg/día (2 tomas) |
| Topiramato, Topamax® (25, 50, 100, 200) | 25-50mg cada 24 h | 25-50 mg/día cada semana | 200-800 mg/día (2 tomas) |
| Gabapentina, Neurontin® (100, 300, 400, 600, 800) | 300-400mg cada 24h (1.er día)-cada 12 h (2.° día)-cada 8 h (3.er día) | 300-400 mg/día cada 1-3 días | 900-3.600 mg/día (2-3 tomas) |
| Oxcarbazepina, Trileptal® (150, 300, 600) | 150-300mg cada 12 h | 600 mg/día cada semana | 600-2.400 mg/día (2 tomas) |
| Levetiracetam, Keppra® (250, 500, 1.000) | 250-500mg cada 12 h | 500-1.000 mg/día cada semana | 1.000-3.000 mg/día (2 tomas) |
| Zonisamida, Zonegran® (25, 50, 100) | 25-50mg cada 24 h | 50-100 mg/día cada semana | 100-500 mg/día (2 tomas) |
| 3.ª generación | |||
| Lacosamida, Vimpat®a (50, 100, 150, 200) | 50mg cada 12 h | 100 mg/día cada semana | 200-400 mg/día (2 tomas) |
| Eslicarbazepina, Zebinix® (800) | 400mg cada 24 h | 400 mg/día cada 1-2 semanas | 400-1.200 mg/día (1 toma) |
| Retigabina, Trobalt® (50, 100, 200, 400) | 100mg cada 8 h | 100-150 mg/día cada semana | 600-1.200 mg/día (3 tomas) |
| Rufinamida, Inovelon® (100, 200, 400) | 100-400mg cada 24 h | 200-400 mg/día cada 2 días | 1.200-4.800 mg/día (2 tomas) |
| Perampanel, Fycompa® (2, 4, 6, 8, 10, 12) | 2mg cada 24 h | 2 mg/día cada semana | 6-12 mg/día (1 toma) |
| Brivaracetam, Briviact® (10, 25, 50, 75, 100) | 25mg cada 12 h | No requerida (la dosis terapéutica se alcanza desde el día 1) | 50-200 mg/día (2 tomas) |
VPA: ácido valproico.
Dosis de carga: 200mg en 15min (oral o intravenosa). Dosis de mantenimiento: 200 mg/día (2 tomas), 12 h después de la dosis de carga.
Adaptada con permiso de Fernández Alonso, 201327.
Las CEs son un fenómeno relativamente frecuente en el ámbito de los CP. Por ello hemos estimado necesario, vinculado a la venidera creación de la Unidad de CP de nuestro centro de NRHB, elaborar este modelo de guía de abordaje de las CEs en CP, el cual aparte de seleccionar de forma apropiada a los pacientes candidatos a recibir CP, resulta crucial de cara a conseguir un control sintomático óptimo de las CEs y soslayar el distrés, el sufrimiento y el dolor vital de estos enfermos y sus familiares, para de esta forma alcanzar un nivel mínimo de bienestar que les haga más llevadera esta etapa final de la vida. Tras una búsqueda bibliográfica exhaustiva, y atendiendo a las características de estos pacientes, se aconseja usar FAEs con presentación vía parenteral (preferiblemente iv) y un perfil bajo de interacciones. DZP y/o MDZ serían los más adecuados para la etapa aguda, y LEV, VPA y/o LCM para los casos refractarios a lo previo que precisen una acción sintomática antiepiléptica más efectiva y/o como tratamiento a largo plazo. Siempre teniendo en mente, que dichas sugerencias deben considerarse como una guía de enfoque global, debiendo adaptarse de forma holística e interdisciplinar, a la naturaleza intrínseca de cada caso clínico en particular, sin desatender las prioridades y deseos del paciente y/o su familia. Por el momento, carecemos de un documento de consenso en relación con esta disyuntiva aprobado a nivel nacional. Así pues, se requieren ensayos clínicos controlados, aleatorizados, bien diseñados, que incluyan muestras amplias de pacientes subsidiarios de CP, para la redacción de dicho documento que permita recomendar con un mayor NE y de forma generalizada, la utilización adecuada, racional y efectiva de FAEs en este ámbito médico-asistencial altamente delicado y complejo.
LimitacionesCon respecto al NE para actuaciones terapéuticas, hemos incluido una tabla inicial aclaratoria, precisando posteriormente en cada actuación el NE y/o el grado de las recomendaciones existentes al respecto, siempre teniendo en mente que el principal problema que concierne a la confección de estas guías es la inexistencia de estudios suficientes como para poder precisar con exactitud el NE de cada intervención en este grupo específico de población, por lo que será un tema pendiente que deberá ser abordado en futuras actualizaciones de esta versión inicial de GPC, conforme vayan apareciendo estudios adecuadamente realizados al respecto.
FinanciaciónNuestro trabajo ha sido redactado libremente, sin financiación por ninguna empresa o entidad pública o privada.
Conflicto de interesesTodos los autores firmantes han aprobado la presentación de este manuscrito. No existen conflictos de intereses.
A todos los pacientes y trabajadores de la Clínica San Vicente.

