



Mucopolysaccharidoses (MPS) are a group of inherited disorders due to lysosomal enzyme deficiencies. The aims of this study are to describe the neuroimaging findings in children evaluated in our hospital with this diagnosis, looking for a possible correlation of these alterations with the type of MPS and clinical severity, and finally to compare these findings with those previously reported.
Material and methodsWe retrospectively analysed the medical records of 19 patients who had been diagnosed with MPS between 1992 and 2010: 7 had type I (5 with Hurler syndrome and 2 with Hurler–Scheie syndrome), 10 had type II or Hunter syndrome (4 with the severe form and 6 with the mild form), 1 had type III or Sanfilippo syndrome and 1 had type VI or Maroteaux–Lamy syndrome. We assessed the brain neuroimaging studies: computed axial tomography (CAT) in 5 patients, and magnetic resonance imaging (MRI) in 15.
ResultsWe observed a broad spectrum of neuroimaging anomalies. In CAT: mega cisterna magna (3/5, 60%). In brain MRI: dilated Virchow–Robin perivascular spaces (11/15, 73%), white matter abnormalities (11/15, 73%), and ventriculomegaly (5/15, 33%).
ConclusionsAbnormal findings in neuroimaging studies are frequent in MPS (dilated Virchow–Robin perivascular spaces, white matter abnormalities and ventriculomegaly). Thus, given these abnormalities we should be aware of this possible diagnosis, particularly when typical signs and symptoms are present. However, we did not find a correlation between these findings and either any specific type of MPS or clinical severity.
Las mucopolisacaridosis (MPS) son un grupo de enfermedades hereditarias de depósito lisosomal. El objetivo de esta revisión es describir las alteraciones neurorradiológicas en los niños evaluados en nuestro hospital con este diagnóstico, buscar la posible correlación de estas alteraciones con el tipo de MPS y con la gravedad clínica, y comparar nuestros hallazgos con lo descrito en la literatura.
Material y métodosRevisamos retrospectivamente las historias clínicas de 19 pacientes diagnosticados de MPS en el periodo 1992–2010: 7 tipo I (5 con síndrome de Hurler y 2 con Hurler–Scheie), 10 tipo II o síndrome de Hunter (4 con la forma grave y 6 con la moderada), 1 tipo III o síndrome de Sanfilippo y 1 tipo VI o síndrome de Maroteaux–Lamy. Se analizaron las pruebas de neuroimagen: tomografía computarizada (TC) en 5 pacientes y resonancia magnética craneal (RMC) en 15.
ResultadosEncontramos un amplio espectro de alteraciones radiológicas. En la TC destaca la megacisterna magna (3/5, 60%); en la RMC el aumento de los espacios perivasculares (11/15, 73%), la alteración parcheada de la sustancia blanca (SB) (11/15, 73%) y la ventriculomegalia (5/15, 33%).
ConclusionesAlgunas anomalías neurorradiológicas son frecuentes en las MPS (aumento de los espacios perivasculares, alteraciones de la SB, ventriculomegalia), por lo que ante estos hallazgos debemos investigar esta posibilidad diagnóstica, especialmente en pacientes con clínica compatible. No hemos hallado datos específicos de cada tipo de MPS, ni relación de estas alteraciones radiológicas con la gravedad de la forma clínica.
Mucopolysaccharidoses (MPS) are a group of congenital errors of metabolism characterised by the deficiency of one of the lysosomal enzymes catalysing the degradation of glycosaminoglycans (GAG) or mucopolysaccharides. This deficiency leads to abnormal accumulation of GAG in the lysosomes, and to it being excessively excreted in urine. There are 7 different types of MPS and their overall prevalence is estimated at 1 in 22500 individuals. These diseases are multisystemic, degenerative, chronic and progressive, and entail a number of disorders, both physical and mental.1,2 They are usually difficult to detect in newborns (unless other family members are affected) since these children are apparently normal at birth. They gradually develop a number of phenotypic abnormalities which include distinctive coarse facial features, changes in the skeletal system (dysostosis multiplex), small size, contractures, cardiovascular disorders (valvulopathy, myocardiopathy), visceromegalies, inguinal and umbilical hernias, skin infiltration disorders, corneal opacity, and hearing impairment.2–4 Neurological manifestations may differ considerably depending on the type of MPS. Progressive cognitive impairment is very frequent in severe MPS types I, II, and VII, and also in type III.5–7 Patients with neurodegeneration are extremely disabled, with death usually occurring in the first or second decade. Patients with milder forms of the disease may reach adolescence and adulthood, but high rates of morbidity are common.5 All types of MPS are inherited by means of recessive autosomal transmission, except for MPS type II or Hunter syndrome, which is an X-linked recessive disease. MPS is diagnosed based on quantitative analysis of GAG levels in the patient's urine. In addition, an enzymatic study specific to the type of MPS and a molecular study are also performed. The treatment of MPS is a complex process.5 Haematopoietic stem cell transplantation has been proven to be effective treatment for some forms of MPS. This is currently the treatment of choice for some patients with severe MPS I; however, morbidity and mortality rates are very high.2 Enzyme replacement therapy has been proven effective for MPS I, II and VI. For all types of MPS, the complications that arise as the disease progresses must be treated correctly.2,5,6
The first neuroradiological studies to evaluate MPS were performed using computed tomography (CT), which revealed non-specific data suggestive of these diseases. Examples of such findings were as follows: hypodense areas in the white matter and dilation of the ventricles and subarachnoid spaces. Later studies showed that brain MRI is more sensitive than CT and offers more detailed information about changes in the central nervous system.8
The purpose of this study is to describe neuropathological alterations in children with any type of MPS who were attended in our hospital. We will search for potential correlations between these changes and the type or severity of MPS, and compare our results with those described in the literature.
Patients and methodsWe retrospectively analysed the medical records of 19 patients who were either diagnosed with MPS or examined for that illness in our department between 1992 and 2010. Initial diagnosis was performed by confirming high urinary GAG excretion and measuring the activity of the corresponding deficient enzyme. Whenever possible, a genetic study was also completed. We classified patients as having different types of MPS according to results from the above studies. In addition, patients may present severe or mild forms of MPS, with poorer or better prognoses, depending on the age at onset of symptoms and the neurological course of the disease. The 19 patients under study were subdivided as follows: 7 with type I (5 had severe MPS or Hurler syndrome and 2 had moderate MPS or Hurler–Scheie syndrome); 10 with type II or Hunter syndrome (4 patients had a severe form and 6 had a moderate form of MPS), 1 type III or Sanfilippo; and 1 type VI or Maroteaux–Lamy syndrome.
The first 4 patients in our series were analysed using brain CT as their only neuroimaging test. These 4 had type II MPS or Hunter syndrome (3 severe cases and 1 moderate case). The other 15 patients were assessed by brain MRI: 5 had Hurler, 2 Hurler–Scheie, 1 severe Hunter, 5 moderate Hunter, 1 Sanfilippo, and 1 Maroteaux–Lamy. One of the patients with Hurler–Scheie syndrome had a brain CT done as an imaging test.
When performing the brain MRI, we used T1 and T2-weighted images along the sagittal and axial planes. We mainly noted the presence or absence of ventriculomegaly or, where applicable, hydrocephalus, patchy white matter changes, increase in perivascular Virchow–Robin spaces (VRS), corpus callosum lesions, cortical and subcortical atrophy, and alterations of the posterior cavity. Images were assessed by expert radiologists who were not informed regarding patients’ neurological conditions. For some of the cases, they were aware of the type of MPS in question. Of the 15 patients who underwent a brain MRI examination, 5 had two or more imaging tests during the follow-up period. The remaining patients underwent a single test, usually performed during the first weeks following the diagnosis or suspected diagnosis.
Once patients were diagnosed, they were assessed clinically on a six-month or yearly basis by the appropriate paediatric sub-specialty (paediatric neurology, otorhinolaryngology, traumatology, cardiology, ophthalmology, etc.) In the past decade, patients monitored by our centre during the entire course of the disease also underwent neuropsychological assessment according to the protocol recommended for these types of diseases.9
ResultsBoth brain CT scans and brain MRIs showed neuroimaging abnormalities in all patients in our series. In the 5 patients who had a CT scan, the most frequent findings were mega cisterna magna, which was present in 3 of the cases (60%); hypodense white matter images (in 2 patients), septum pellucidum cyst and partial agenesis of the corpus callosum.
The most frequent MRI findings were increase in VRS (11/15, 73%); patchy changes in white matter (11/15, 73%); and ventriculomegaly (5/15, 33%). We found subcortical atrophy in 3 of the patients (1 patient with Hurler–Scheie syndrome, 1 with Hurler syndrome, and 1 with Sanfilippo syndrome). Some of the patients presented alterations of the posterior cranial fossa, including slight reduction of the anterior and posterior subarachnoid space in the bulbomedullary union in the patient with MPS VI; megacisterna magna in 2 children (1 with moderate Hunter syndrome and the other with Hurler syndrome); and a retrocerebellar cyst. We found alterations of the corpus callosum in 5 patients, comprising focal lesions in a patient with Hurler syndrome, increase in VRS in 2 children with Hunter syndrome (1 moderate, the other severe), and atrophy of the corpus callosum in the patient with MPS type III and1 patient with moderate Hunter syndrome.
During follow-up, 5 patients had a cervical MRI and 5 patients had a complete or lumbosacral spinal MRI. We found changes in the high cervical area in 5 patients: 2 with Hurler–Scheie syndrome, 1 with severe Hunter syndrome, 1 with moderate Hunter syndrome, and 1 with Maroteaux–Lamy syndrome. In one of the girls with Hurler–Scheie syndrome we found an increase of the retro-odontoid soft tissues and correction of physiological cervical lordosis, with decreased width of the spinal canal in the high cervical area. In another patient with moderate type II MPS, we discovered a slight thickening of the soft tissues behind the odontoid apophysis, with no significant decrease of the width of the spinal canal. Other isolated findings were deformity of the third cervical vertebral body (in a child with severe Hunter syndrome) and hypoplasia of the odontoid apophysis (in the patient with MPS VI and in 1 girl with Hurler–Scheie syndrome). In the 5 children with an MRI study of the lumbosacral spine, we found anterior and posterior wedging or deformity of the first or second lumbar vertebra.
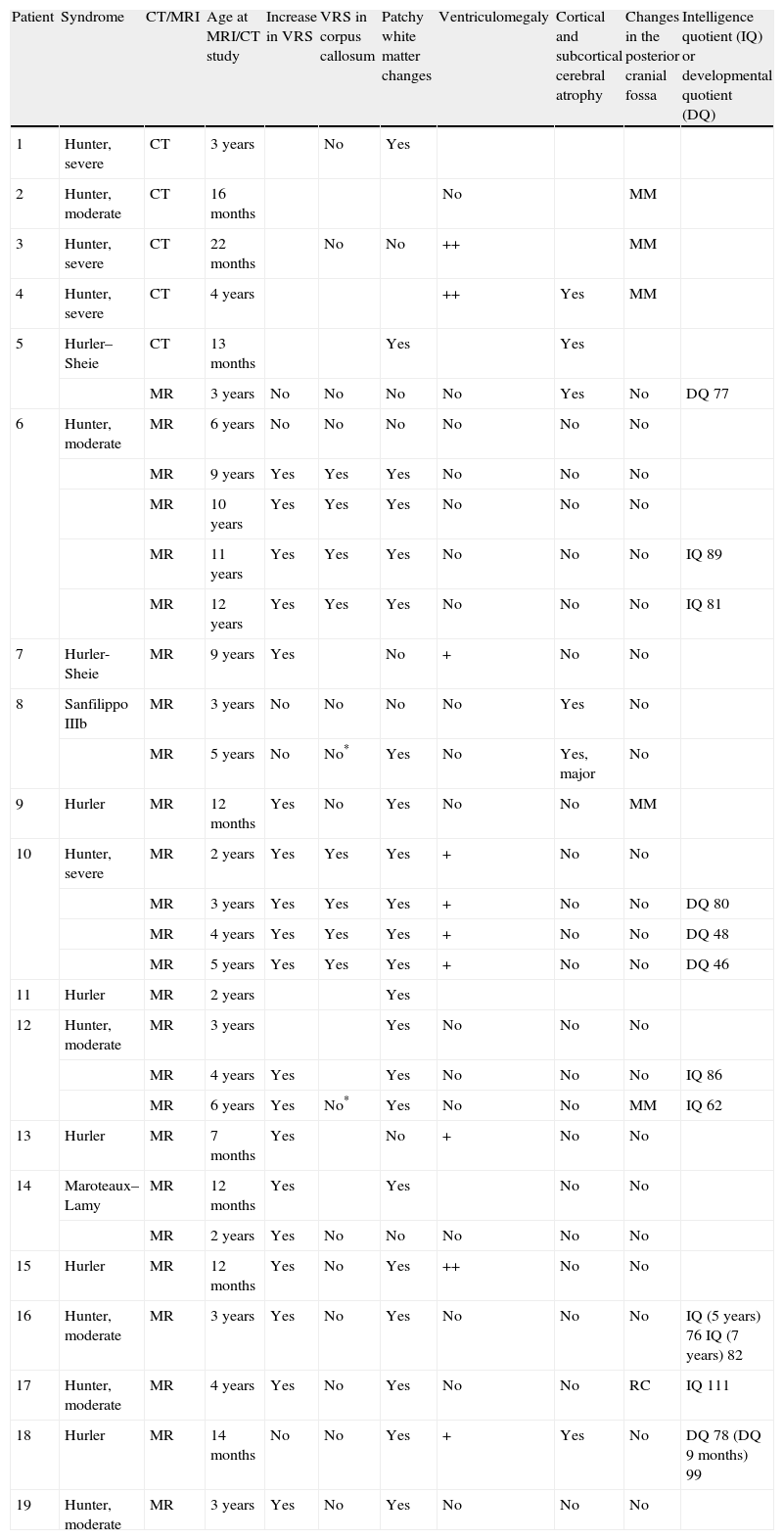
In cases with MPS I or II (the most frequent in our series), we found no link between neuroradiological changes and specific type of MPS, or between changes and disease severity. In the last patients studied in our centre, those with a complete neuropsychological study, we observed no correlations between radiological changes the patient's level of cognitive development. The study was performed using the Weschler scales10,11 for children older than 3 years, and the Batelle developmental scale12 in younger or significantly impaired children (Table 1).
Radiological findings from patients included in the study.
| Patient | Syndrome | CT/MRI | Age at MRI/CT study | Increase in VRS | VRS in corpus callosum | Patchy white matter changes | Ventriculomegaly | Cortical and subcortical cerebral atrophy | Changes in the posterior cranial fossa | Intelligence quotient (IQ) or developmental quotient (DQ) |
| 1 | Hunter, severe | CT | 3 years | No | Yes | |||||
| 2 | Hunter, moderate | CT | 16 months | No | MM | |||||
| 3 | Hunter, severe | CT | 22 months | No | No | ++ | MM | |||
| 4 | Hunter, severe | CT | 4 years | ++ | Yes | MM | ||||
| 5 | Hurler–Sheie | CT | 13 months | Yes | Yes | |||||
| MR | 3 years | No | No | No | No | Yes | No | DQ 77 | ||
| 6 | Hunter, moderate | MR | 6 years | No | No | No | No | No | No | |
| MR | 9 years | Yes | Yes | Yes | No | No | No | |||
| MR | 10 years | Yes | Yes | Yes | No | No | No | |||
| MR | 11 years | Yes | Yes | Yes | No | No | No | IQ 89 | ||
| MR | 12 years | Yes | Yes | Yes | No | No | No | IQ 81 | ||
| 7 | Hurler-Sheie | MR | 9 years | Yes | No | + | No | No | ||
| 8 | Sanfilippo IIIb | MR | 3 years | No | No | No | No | Yes | No | |
| MR | 5 years | No | No* | Yes | No | Yes, major | No | |||
| 9 | Hurler | MR | 12 months | Yes | No | Yes | No | No | MM | |
| 10 | Hunter, severe | MR | 2 years | Yes | Yes | Yes | + | No | No | |
| MR | 3 years | Yes | Yes | Yes | + | No | No | DQ 80 | ||
| MR | 4 years | Yes | Yes | Yes | + | No | No | DQ 48 | ||
| MR | 5 years | Yes | Yes | Yes | + | No | No | DQ 46 | ||
| 11 | Hurler | MR | 2 years | Yes | ||||||
| 12 | Hunter, moderate | MR | 3 years | Yes | No | No | No | |||
| MR | 4 years | Yes | Yes | No | No | No | IQ 86 | |||
| MR | 6 years | Yes | No* | Yes | No | No | MM | IQ 62 | ||
| 13 | Hurler | MR | 7 months | Yes | No | + | No | No | ||
| 14 | Maroteaux–Lamy | MR | 12 months | Yes | Yes | No | No | |||
| MR | 2 years | Yes | No | No | No | No | No | |||
| 15 | Hurler | MR | 12 months | Yes | No | Yes | ++ | No | No | |
| 16 | Hunter, moderate | MR | 3 years | Yes | No | Yes | No | No | No | IQ (5 years) 76 IQ (7 years) 82 |
| 17 | Hunter, moderate | MR | 4 years | Yes | No | Yes | No | No | RC | IQ 111 |
| 18 | Hurler | MR | 14 months | No | No | Yes | + | Yes | No | DQ 78 (DQ 9 months) 99 |
| 19 | Hunter, moderate | MR | 3 years | Yes | No | Yes | No | No | No |
VRS: Virchow–Robin spaces; MM: mega cisterna magna; RC: retrocerebellar cyst; +: mild; ++: moderate to severe.
Slight atrophy was described in these cases.
MPS are a group of heterogeneous diseases that produce somatic effects and, in some cases, neurodegeneration. Suspected diagnosis is based on characteristic clinical signs and symptoms, family history, and high levels of GAG excretion in urine. Diagnosis is then confirmed with the aid of an enzyme and genetic study.2,5 MRI is a useful technique for evaluating brain and spinal changes, including changes in white matter, hydrocephalus, and spinal cord compression.3,13,14 Neuroradiological changes in cerebral parenchyma in MPS have been described before, although not much literature is available on the subject. These conditions have a low prevalence overall, and brain MRI became a widely used testing method only recently.8 It is therefore difficult to conclude whether or not there is a link between these changes and the type of MPS, or the severity of its clinical form.
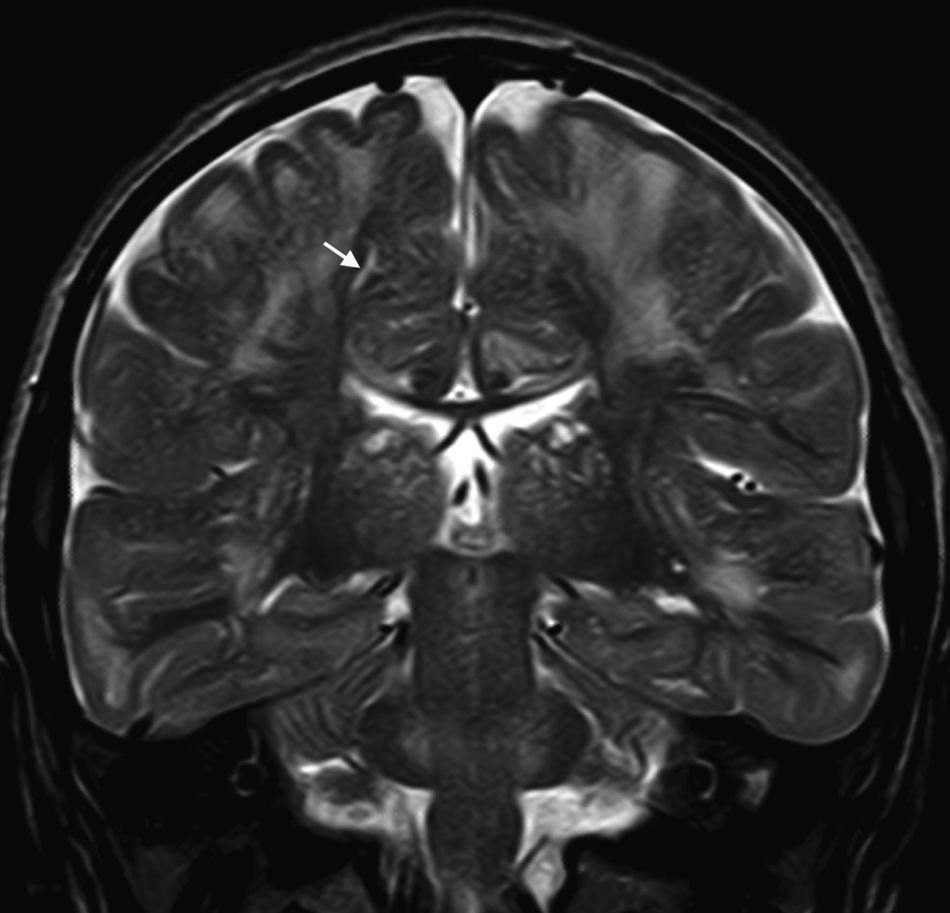
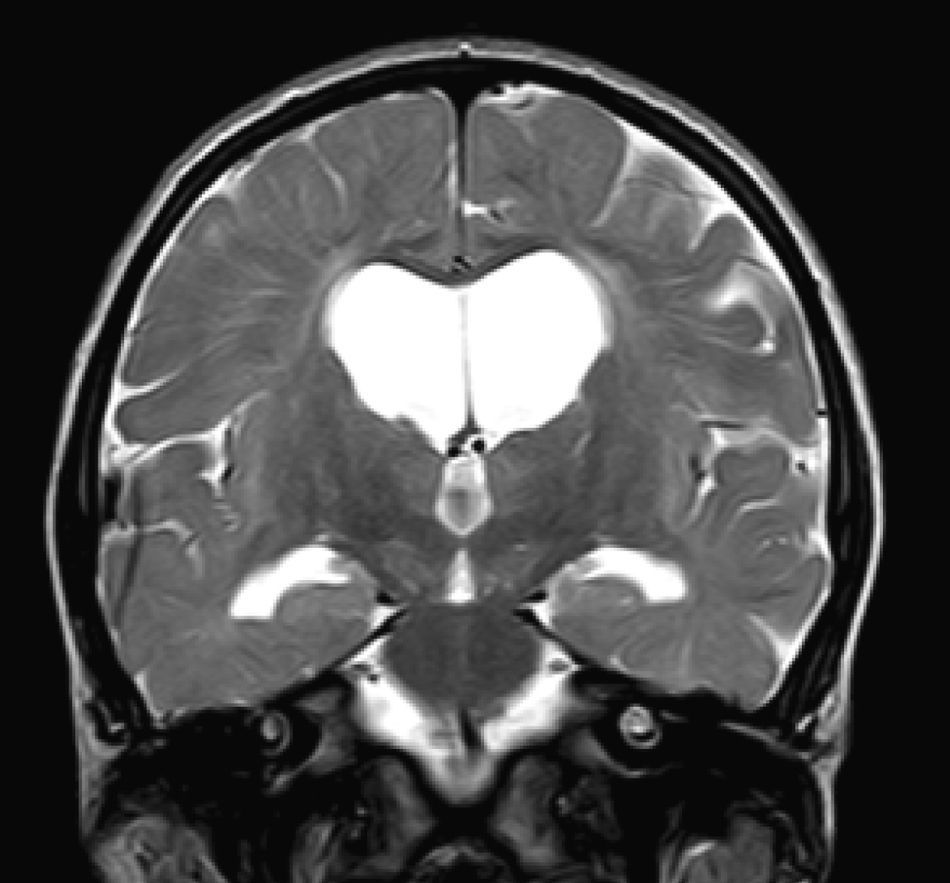
In our series, the dominant CT finding was mega cisterna magna (3/5, 60%). The most frequent findings from brain MRI are increase in VRS (11/15, 73%), patchy white matter changes (11/15, 73%) and ventriculomegaly (5/15, 33%). This coincides with that published in the literature.3,8,13–17 Seto et al.13 observed that increases in VRS are more common and severe in MPS II, while patchy white matter changes are the most common in MPS I and II. Ventriculomegaly is most frequently found in types I and II. We found an increase in VRS in MPS I, II, and VI (Fig. 1) as did the cited author, as well as patchy white matter lesions in MPS types I, II, and VI and ventriculomegaly in MPS types I and II (Fig. 2). The neuroradiological signs found in our group of patients are therefore quite characteristic, but none is specific to a single type of MPS.15–17
. Note the hyperintense signals in T2 on the supratentorial white matter and marked dilation of perivascular Virchow–Robin spaces (arrow) in a patient with Hunter syndrome.")
. We identified supratentorial ventriculomegaly and dilation of perivascular Virchow–Robin spaces in a patient with severe Hunter syndrome aged 5 and a half.")
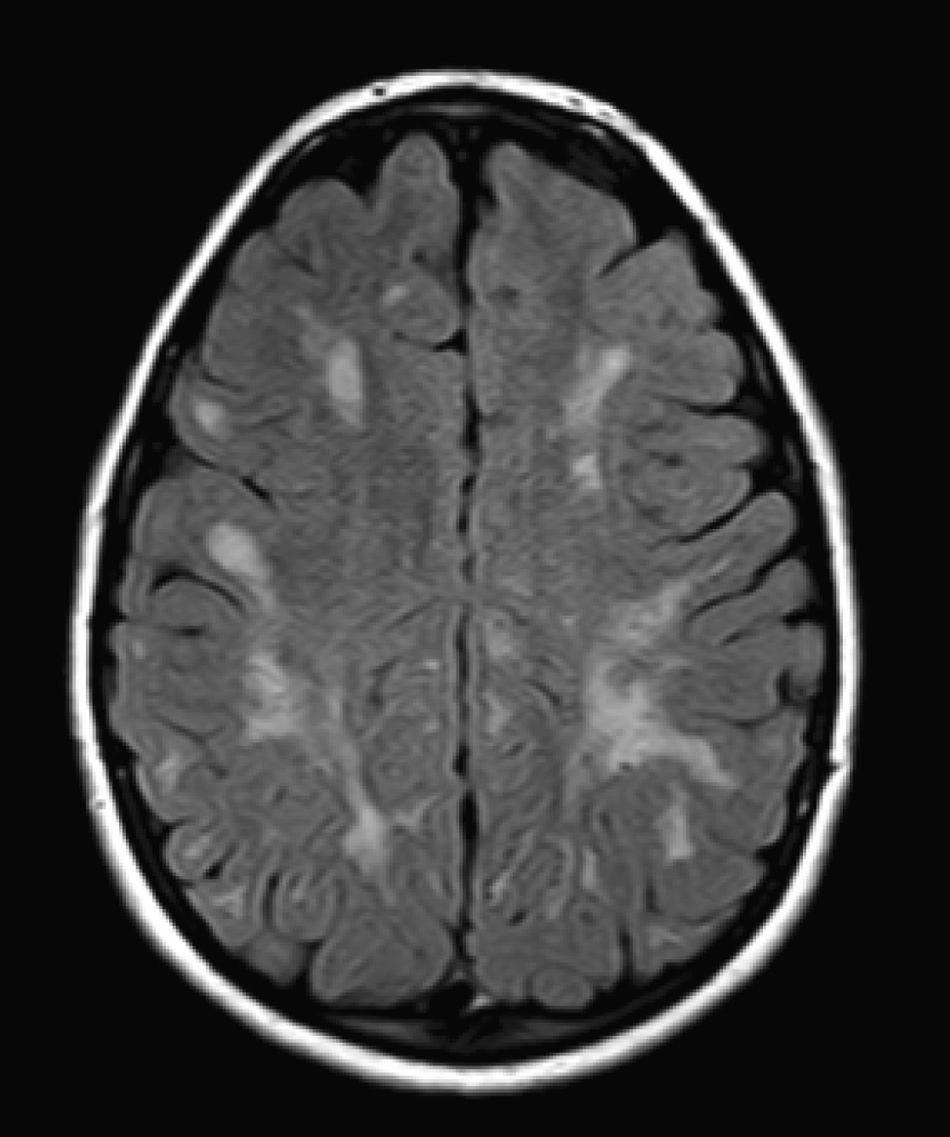
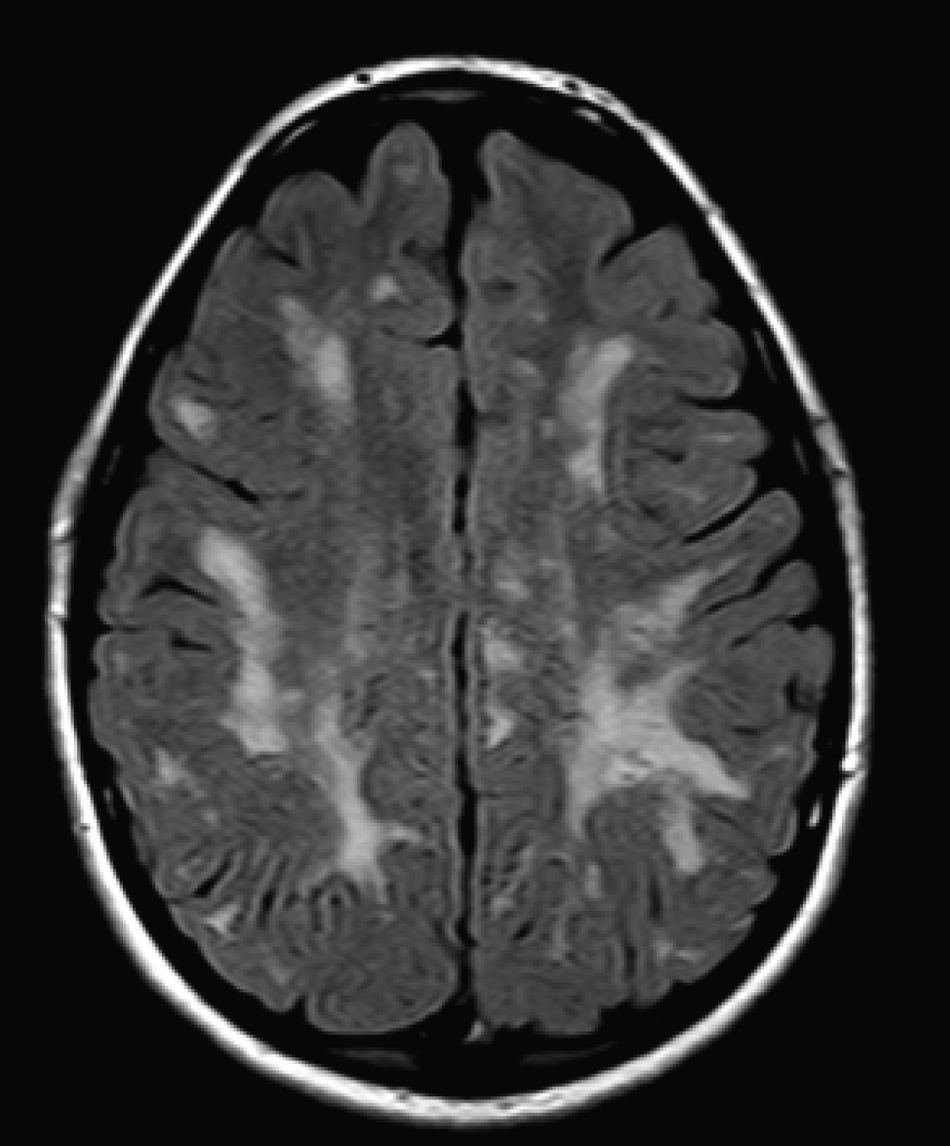
We did not find any correlations between brain MRI findings and the type of MPS or the severity of its clinical form. Neither of the 2 females with Hurler–Scheie syndrome showed patchy white matter changes, and this was also true of 1 of the 4 patients with Hurler syndrome. We observed increase in VRS in 5 males with moderate Hunter syndrome and in another patient with a severe form of the disease. This means that the brain alterations described in our patients (increase in VRS, patchy white matter changes, ventriculomegaly, etc.) may be independent from the severity of the condition. However, other studies point to a direct link between the extent of these neuroradiological alterations and the severity of the disease.13 Some studies in particular found a correlation between cognitive level and white matter changes (but not with other neuroimaging changes).18 Others link the extent of white matter changes with the severity of cognitive decline.19 Our series contained a patient with moderate Hunter syndrome who had been treated for more than 4 years with weekly enzyme substitution treatment and radiological follow-up with a yearly brain MRI from time of diagnosis up to the present. This patient's most recent radiological study showed progression of the patchy white matter lesions (Figs. 3 and 4). However, the neuropsychological study did not show any significant decrease (greater than 1 typical deviation) in total IQ. In fact, the patient showed improvement in areas such as auditory processing, expressive language, and verbal memory, plus fewer motor differences compared to a previous study. Nevertheless, some recent studies seem to show that enzyme substitution treatment is followed by improvement in radiological images (especially white matter lesions) in patients with mild type I MPS.20 Others report improvement or stabilisation of neuroimaging alterations in patients with type I and II MPS.21


Increases in retro-odontoid soft tissue with a decrease in the diameter of the spinal canal (which can lead to symptoms of spinal cord compression) as well as other spinal changes have been described in MPS.1,14,22 In our series, we also found MRI changes in the cervical area and in other parts of the spinal column. This highlights the importance of performing these tests in patients with MPS, especially if neurological signs are compatible with such changes.
Our series has obvious limitations. Serial MRI studies were only completed in 5 patients, and the ages at which brain MRIs were performed are heterogeneous. In addition, the number of study subjects is low, and the results we obtained are therefore merely descriptive. A much larger sample would be required in order to compare and obtain more reliable results. Evaluating progressive neuroradiological changes and studying correlations between such findings and cognitive decline are recommendable in this group of diseases. This is likely to be an easier task at present, since MR technology is now widely available. Radiological changes found in MPS are not specific, but they can be suggestive of this group of diseases. A possible diagnosis of MPS should therefore be studied in cases of children with hernia, joint stiffness, visceromegaly, hearing loss, intellectual disability, bone disorders, or other typical symptoms together with a compatible brain MRI image.
Conflicts of interestThe authors declare that there are no conflicts of interest.
Please cite this article as: Calleja Gero ML, et al. Hallazgos neurorradiológicos en una serie de pacientes con mucopolisacaridosis. Neurología. 2012;27:407–13.
