






Se realizó un estudio epidemiológico molecular en una población de 9.422 donantes de sangre de la provincia de Corrientes (noreste de Argentina), con el fin de determinar la prevalencia del virus linfotrópico T del humano tipos 1 y 2 (human T-cell lymphotropic virus: HTLV-1/2), de identificar filogenéticamente a los subtipos/subgrupos de HTLV-1 y 2 encontrados y de realizar el análisis de mutaciones. Sobre la base de los resultados obtenidos, se demostró que tanto el HTLV-1 como el HTLV-2 se encuentran circulando en una población de bajo riesgo de Corrientes, si bien con una prevalencia similar a las de áreas no endémicas. Los estudios filogenéticos identificaron al subtipo Cosmopolita subgrupo Transcontinental (Aa) del HTLV-1 y al subtipo b del HTLV-2. Los donantes infectados no manifestaron antecedentes de riesgo tales como transfusiones, uso de drogas inyectables ni parejas sexuales de riesgo o seropositivas para HTLV-1/2. Estos resultados indican que estos virus fueron transmitidos de madre a hijo, posiblemente de generación en generación, y que estas cepas fueron introducidas en la población caucásica de esta región a partir de ascendientes originarios de áreas endémicas del país o por contacto producido tiempo atrás con individuos infectados de otros países. Nuestros resultados demuestran por primera vez la presencia de HTLV-1 y HTLV-2 en la provincia de Corrientes. Y si bien se puede considerar a esta provincia como área no endémica, se destaca la necesidad de incluir a estos retrovirus en un programa nacional de salud pública, con el fin de contar con profesionales capacitados para realizar su diagnóstico y brindar la información necesaria en relación con la atención primaria y el seguimiento de los pacientes.
A molecular epidemiological study was conducted in a population of 9422 blood donors in the province of Corrientes, Northeastern Argentina, to determine the prevalence of Human T-cell lymphotropic virus types 1 and 2 (HTLV-1/2), the phylogenetic identification of HTLV-1 and 2 subtypes/subgroups and perform a mutation analysis. Based on the results obtained, it was shown that both HTLV-1 and HTLV-2 are circulating in a low-risk population of Corrientes, although with a similar prevalence to that of non-endemic areas. Phylogenetic studies identified the HTLV-1 Cosmopolitan subtype Transcontinental subgroup (Aa), and the HTLV-2 subtype b. Infected donors reported neither a history of risk factors such as transfusions, intravenous drug use, nor risky or HTLV-1/2 seropositive sexual partners. These results suggest that these viruses were transmitted from mother to child, possibly from generation to generation, and that these strains were introduced into the Caucasian population of this region from ancestors originating from endemic areas of the country either from or through contact with individuals from other countries years ago. Our results demonstrate for the first time the presence of HTLV-1 and HTLV-2 in the province of Corrientes. Moreover, although the province can be considered a non-endemic area, the need to include these retroviruses in a national Public Health program is highlighted, in order to have qualified professionals duly trained to make their diagnosis and provide the necessary information in relation to primary care and patient follow-up.
El virus linfotrópico T humano 1 (human T-cell lymphotropic virus: HTLV-1), primer oncorretrovirus humano, descubierto en 1980, es el agente etiológico de la leucemia/linfoma de células T del adulto y de la mielopatía asociada al HTLV-1/paraparesia espástica tropical. También está relacionado con otras enfermedades inflamatorias y autoinmunitarias, como la artropatía inflamatoria crónica, el síndrome de Sjögrens, la polimiositis, la uveítis, la alveolitis y la dermatitis infecciosa11. Este retrovirus es endémico en distintas partes del mundo y también en el noroeste argentino (Jujuy y Salta), donde ambas enfermedades, la leucemia/linfoma de células T del adulto y la mielopatía asociada al HTLV-1/paraparesia espástica tropical, son endémicas4,12,19,22.
El HTLV-2, virus linfotrópico T humano 2 (human T-cell lymphotropic virus-2: HTLV-2), oncorretrovirus humano aislado en 1982, infecta preferentemente a los linfocitos T CD8+ (a diferencia del HTLV-1 y del virus de la inmunodeficiencia humana 1, o HIV-1, que presentan tropismo por los linfocitos T CD4+), y hasta el presente, no se lo ha asociado con ninguna enfermedad específica14.
El HTLV-2 es naturalmente endémico en algunas poblaciones de aborígenes del Nuevo Mundo, incluyendo los tobas y wichis de Argentina, y en tribus de África central3,14. Ambos retrovirus se encuentran en poblaciones de alto riesgo y de bajo riesgo, con distintas cifras de prevalencia, que varían en relación con el grupo, el año de estudio y la región geográfica10,14. En donantes de sangre de áreas no endémicas, las cifras de prevalencia varían de 0,01 a 0,07%10,17. El HTLV-1 y el HTLV-2, de manera similar al HIV-1, se transmiten por vía sexual, parenteral y de madre a hijo (principalmente por la lactancia)27. El objetivo de este trabajo es aportar mayores conocimientos sobre el HTLV-1/2 en Argentina, evaluando por primera vez la situación de la infección en la provincia de Corrientes (nordeste de Argentina).
Materiales y métodosSe estudiaron muestras de donantes de sangre consecutivos, aceptados entre enero de 2007 y diciembre de 2010 por el Servicio de Hemoterapia del Instituto de Cardiología de Corrientes Juana F. Cabral (ciudad de Corrientes). Además, se recolectaron los datos sociodemográficos y los antecedentes de transfusión y donaciones de sangre. Las muestras de los individuos incluidos en el estudio fueron identificadas e ingresadas según la Ley Nacional de Sida de nuestro país. Se mantuvo la confidencialidad de toda la información, según lo indican las pautas de la legislación nacional e internacional vigente. Se siguieron estrictamente las pautas éticas internacionales relacionadas con los estudios epidemiológicos y clínicos (Pautas internacionales para la evaluación ética de los estudios epidemiológicos, CIOMS, Ginebra, 1991).
Diagnóstico de la infección por HTLV-1/2Las muestras de los donantes fueron procesadas por duplicado con diferentes equipos comerciales de ELISA: Vironostika HTLV-1/2 (BioMérieux, Holanda), Detect-HTLV, Adaltis (Montreal, Canadá); bioelisa HTLV, Biokit (Barcelona, España) o HTLV-I/HTLV-II ELISA 4.0, MP Diagnostics (Santa Ana, California, EE. UU.), según el momento del estudio. A su vez, todas las muestras repetidamente reactivas fueron analizadas por aglutinación de partículas de gelatina (Serodia-HTLV-I, Fujirebio, Japón). Los análisis y la interpretación de los resultados se realizaron siguiendo de manera estricta las instrucciones de los fabricantes. Las muestras que se mantuvieron como reactivas a lo largo de reiteradas pruebas de tamizaje fueron analizadas en el Instituto de Investigaciones Biomédicas en Retrovirus y Sida (INBIRS) por la técnica de Western blot (WB) (HTLV Blot 2.4, Genelabs Diagnostics, Singapur Science Park, Singapur).
Análisis filogenéticoLas muestras de sangre entera de los casos seropositivos para HTLV-1/2 por WB fueron descongeladas y a partir de aquellas se realizó la extracción de ADN con un equipo comercial (ADN PuriPrep-S kit, Highway, Inbio, Tandil, Argentina). Para la amplificación del ADN proviral se realizó una nested PCR para la región LTR del HTLV-1 con los cebadores externos 8200LA/3VEXT y los internos 8200LA/3VINT20. Para el LTR del HTLV-2 se utilizaron los cebadores externos BSQF6/BSDR3 e internos BSQF2/BSDR424. El tamaño de los productos finales de amplificación fue de 528 pb y 665 pb para el HTLV-1 y el 2, respectivamente.
Luego de la purificación, se realizó la secuenciación automática utilizando el kit BigDye® Terminator v. 3.1 Cycle Sequencing Kits (Applied BioSystems, Foster City, CA, EE.UU.), en un equipo ABi Prism 3100 Genetic Analizer (Applied Biosystems), abarcando doble marco de lectura. Los cromatogramas se editaron utilizando el programa Sequencher 4.8 (Gene Codes Co, Ann Arbor, MI, EE.UU.). Las secuencias fueron examinadas con el programa BioEdit (Ibis Therapeutics, Carlsbad, CA, EE.UU.) alineadas con el algoritmo ClustalW (University College Dublin, Dublin, Irlanda) y corregidas manualmente13,26. Los cálculos de distancia media y la reconstrucción filogenética por medio de neighbour joining se realizaron utilizando el programa MEGA (Molecular Evolutionary Genetics Analysis) versión 4 para Windows (Center for Evolutionary Medicine and Informatics, Tempe, AZ, EE.UU.)25.
Para el análisis del HTLV-1, las secuencias fueron alineadas junto a 133 muestras de referencia de dicho virus obtenidas de la base de datos de GeneBank, elegidas preferentemente porque pertenecían a nuestro país, a países limítrofes con altas tasas de migración a la Argentina o porque habían sido reportadas en otros países de América del Sur. La secuencia de referencia Mel 5, de origen melanésico y subtipo c, fue utilizada como grupo externo. Una vez alineadas y editadas las secuencias, la base de datos consistió en secuencias de 490 pb correspondientes a la región 3’LTR. La matriz se analizó por máxima verosimilitud.
Para el análisis del HTLV-2, las secuencias estudiadas fueron alineadas junto a 78 secuencias de referencia de dicho virus obtenidas de la base de datos de GeneBank, escogidas por provenir de países limítrofes con una alta tasa de migración a la Argentina, además de secuencias representativas del resto del mundo. Como grupo externo se empleó una secuencia de referencia de STLV-2 (PP1664). Una vez alineadas, las secuencias consistieron en 596 pb correspondientes a la región 3’LTR.
Análisis de mutacionesSe realizó un alineamiento de las secuencias 3’LTR de los aislamientos analizados filogenéticamente con el programa Bioedit, incluyendo la secuencia prototipo ATK-1 para HTLV-1 y NRA para HTLV-2, las cuales se utilizaron para fijar las posiciones nucleotídicas y determinar la presencia de mutaciones correspondientes a cada tipo viral.
Análisis estadísticosPara los estudios epidemiológicos, la prevalencia fue estimada como la proporción de casos positivos confirmados en toda la población evaluada. Los IC 95% fueron calculados asumiendo una distribución binomial. La prevalencia y sus cambios en el tiempo fueron analizados con la prueba de Chi2, estratificados por lugar de residencia, edad y sexo. Los análisis estadísticos fueron realizados utilizando el programa SPSS, versión 11.5.0, 2002 (Chicago, IL, EE. UU).
Para comparar proporciones, se utilizaron tanto las pruebas de Chi2 como exacta de Fisher. En los estudios filogenéticos, el porcentaje de similitud fue obtenido mediante la comparación de la secuencia prototipo y las muestras analizadas, para cada uno de los subtipos (prueba de Kruskal-Wallis). La media estimada fue con un IC 95%.
ResultadosSe estudiaron un total de 9.422 muestras de donantes de sangre de la ciudad de Corrientes entre enero de 2007 y diciembre de 2010. El rango de edad de los donantes fue de 18 a 65 años, con una edad promedio de 36,7 años. Tres muestras resultaron reactivas para HTLV-1/2 por ELISA y aglutinación de partículas; cuando se procedió a la confirmación por WB, una muestra fue positiva para HTLV-1 y 2para HTLV-2. La prevalencia final de HTLV-1/2 fue de 0,032%, con 0,011% para HTLV-1 y 0,021% para HTLV-2.
El donante HTLV-1 positivo era un hombre de 43 años, nacido en el departamento de San Cosme (Corrientes) y residente en la ciudad capital de la provincia de Corrientes. En cuanto a los 2donantes HTLV-2 positivos, uno de ellos era un hombre de 22 años nacido y domiciliado en la ciudad capital de Corrientes; el otro era un hombre de 25 años, nacido y domiciliado en la ciudad correntina de Bella Vista, ubicada a 143km al sur de la capital. Ninguno de los individuos positivos reportó antecedentes de riesgo como transfusiones, uso de drogas ilegales o parejas sexuales de riesgo o seropositivas para HTLV-1/2. Tampoco presentaron coinfecciones con otros agentes de tamizaje obligatorio en Argentina, como HIV, virus de la hepatitis B, virus de la hepatitis C, Treponema pallidum, Trypanosoma cruzi y Brucella sp.
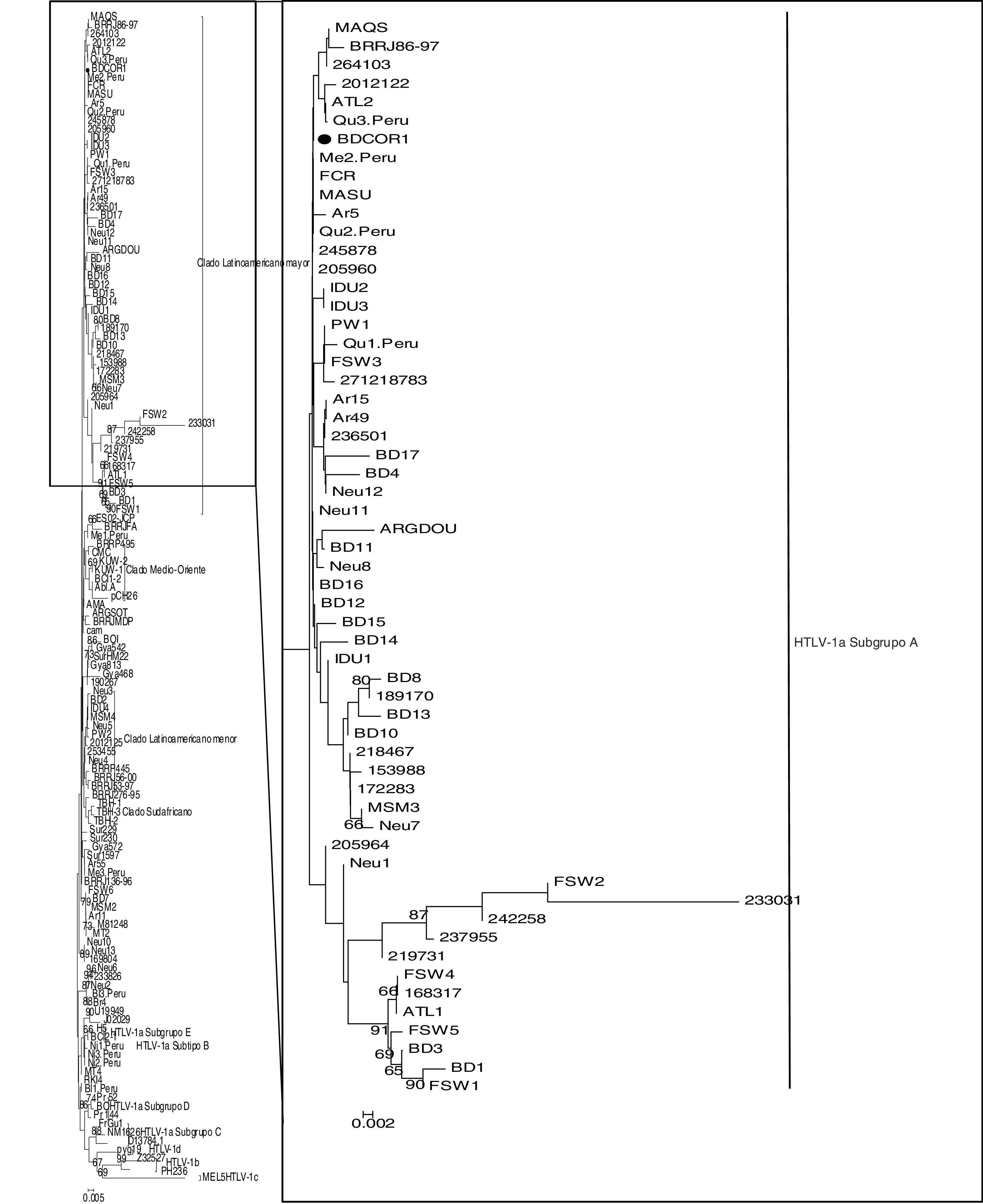
En cuanto al análisis filogenético, el aislamiento HTLV-1 (BDCOR1) se agrupó con el subtipo Cosmopolita (valor de bootstrap: 60%) y el subgrupo Transcontinental (valor de bootstrap: 36%) (fig. 1). La secuencia se agrupó con referencias pertenecientes al grupo latinoamericano mayor, junto con secuencias de donantes de sangre de otras provincias de Argentina, como Buenos Aires (233826 y 264103), Misiones (2012122), Santiago del Estero (245878) y Tucumán (205960), además de donantes amerindios provenientes del noroeste argentino (Ar5), Brasil (MASU, FCR, MAQS, BRRJ86-97) y Perú (Me2.Peru, Qu2.Peru, Qu3.Peru).
, BRRJ86-97 (Brasil), 264103 (Argentina), 2012122 (Argentina), ATL2 (Argentina), Qu3.Peru (Perú), BDCOR1 (Argentina), Me2.Peru (Peru), FCR (Brasil), MASU (Brasil), Ar5 (Argentina), Qu2.Peru (Perú), 245878 (Argentina), 205960 (Argentina), IDU2 (Indonesia), IDU3 (Indonesia), PW1 (Argentina), Qu1.Peru (Peru), FSW3 (Argentina), 271218783 (Argentina), Ar15 (Argentina), Ar49 (Argentina), 236501 (Argentina), BD17 (Argentina), BD4 (Argentina), Neu12 (Argentina), Neu11 (Argentina), ARGDOU (Argentina), BD11 (Argentina), Neu8 (Argentina), BD16 (Argentina), BD12 (Argentina), BD15 (Argentina), BD14 (Argentina), IDU1 (Argentina), BD8 (Argentina), 189170 (Argentina), BD13 (Argentina), BD10 (Argentina), 218467 (Argentina), 153988 (Argentina), 172283 (Argentina), MSM3 (Argentina), Neu7 (Argentina), 205964 (Argentina), Neu1 (Argentina), FSW2 (Argentina), 233031 (Argentina), 242258 (Argentina), 237955 (Argentina), 219731 (Argentina), FSW4 (Argentina), 168317 (Argentina), ATL1 (Argentina), FSW5 (Argentina), BD3 (Argentina), BD1 (Argentina), FSW1 (Argentina), ES02-JCP (Brasil), BRRJFA (Brasil), Me1.Peru (Perú), BRRP495 (Brasil), CMC (Taiwán), KUW-2 (Kuwait), KUW-1 (Kuwait), BCI1-2 (Columbia Británica), Abl.A (Sudáfrica), CH26 (Chile), AMA (Brasil), ARGSOT (Argentina), BRRJMDP (Brasil), cam (Guayana Francesa), BOl (Francia), Gya542 (Guyana), SurHM22 (Surinam), Gya813 (Guyana), Gya468 (Guyana), 190267 (Argentina), Neu3 (Argentina), BD2 (Argentina), IDU4 (Argentina), MSM4 (Argentina), Neu5 (Argentina), PW2 (Argentina), 2012125 (Argentina), 253455 (Argentina), Neu4 (Argentina), BRRP445 (Brasil), BRRJ56-00 (Brasil), BRRJ53-97 (Brasil), BRRJ276-95 (Brasil), TBH-1 (Sudáfrica),TBH-3 (Sudáfrica), TBH-2 (Sudáfrica), Sur229 (Surinam), Sur230 (Surinam), Gya572 (Guyana), Sur1597 (Surinam), Ar55 (Argentina), Me3.Peru (Perú), BRRJ136-96 (Brasil), FSW6 (Argentina), BD7 (Argentina), MSM2 (Argentina), Ar11 (Argentina), M81248 (USA), MT2 (Japón), Neu10 (Argentina), Neu13 (Argentina), 169804 (Argentina), Neu6 (Argentina), 233826 (Argentina), Neu2 (Argentina), Bl3.Peru (Perú), Br4 (Brasil), U19949 (Japón), ATK-1 (Japón), H5 (Japón), BCI2-1 (Canadá), Ni1-3.Peru (Perú), MT4 (Francia), RKI4.Peru (Perú), Bl1.Peru (Perú), Pr52 (Marruecos), BO (Argelia), Pr144 (Marruecos), FrGu1 (Guayana Francesa), NM1626.lac (Guayana Francesa), D13784 (Caribe), pyg19 (África central), ITIS (República Democrática del Congo), PH236 (Gabón), MEL5 (Islas Salomón).")
Árbol de maximum likelihood de 133 secuencias de un fragmento de 490 pb del LTR del HTLV-1. La secuencia correspondiente a la muestra analizada se muestra con ●. La secuencia MEL5 se utilizó como outgroup. Los números en las ramas indican el valor de soporte de cada nodo. El origen geográfico de las secuencias de referencia incluidas en el análisis de arriba hacia abajo son los siguientes: MAQS (Brasil), BRRJ86-97 (Brasil), 264103 (Argentina), 2012122 (Argentina), ATL2 (Argentina), Qu3.Peru (Perú), BDCOR1 (Argentina), Me2.Peru (Peru), FCR (Brasil), MASU (Brasil), Ar5 (Argentina), Qu2.Peru (Perú), 245878 (Argentina), 205960 (Argentina), IDU2 (Indonesia), IDU3 (Indonesia), PW1 (Argentina), Qu1.Peru (Peru), FSW3 (Argentina), 271218783 (Argentina), Ar15 (Argentina), Ar49 (Argentina), 236501 (Argentina), BD17 (Argentina), BD4 (Argentina), Neu12 (Argentina), Neu11 (Argentina), ARGDOU (Argentina), BD11 (Argentina), Neu8 (Argentina), BD16 (Argentina), BD12 (Argentina), BD15 (Argentina), BD14 (Argentina), IDU1 (Argentina), BD8 (Argentina), 189170 (Argentina), BD13 (Argentina), BD10 (Argentina), 218467 (Argentina), 153988 (Argentina), 172283 (Argentina), MSM3 (Argentina), Neu7 (Argentina), 205964 (Argentina), Neu1 (Argentina), FSW2 (Argentina), 233031 (Argentina), 242258 (Argentina), 237955 (Argentina), 219731 (Argentina), FSW4 (Argentina), 168317 (Argentina), ATL1 (Argentina), FSW5 (Argentina), BD3 (Argentina), BD1 (Argentina), FSW1 (Argentina), ES02-JCP (Brasil), BRRJFA (Brasil), Me1.Peru (Perú), BRRP495 (Brasil), CMC (Taiwán), KUW-2 (Kuwait), KUW-1 (Kuwait), BCI1-2 (Columbia Británica), Abl.A (Sudáfrica), CH26 (Chile), AMA (Brasil), ARGSOT (Argentina), BRRJMDP (Brasil), cam (Guayana Francesa), BOl (Francia), Gya542 (Guyana), SurHM22 (Surinam), Gya813 (Guyana), Gya468 (Guyana), 190267 (Argentina), Neu3 (Argentina), BD2 (Argentina), IDU4 (Argentina), MSM4 (Argentina), Neu5 (Argentina), PW2 (Argentina), 2012125 (Argentina), 253455 (Argentina), Neu4 (Argentina), BRRP445 (Brasil), BRRJ56-00 (Brasil), BRRJ53-97 (Brasil), BRRJ276-95 (Brasil), TBH-1 (Sudáfrica),TBH-3 (Sudáfrica), TBH-2 (Sudáfrica), Sur229 (Surinam), Sur230 (Surinam), Gya572 (Guyana), Sur1597 (Surinam), Ar55 (Argentina), Me3.Peru (Perú), BRRJ136-96 (Brasil), FSW6 (Argentina), BD7 (Argentina), MSM2 (Argentina), Ar11 (Argentina), M81248 (USA), MT2 (Japón), Neu10 (Argentina), Neu13 (Argentina), 169804 (Argentina), Neu6 (Argentina), 233826 (Argentina), Neu2 (Argentina), Bl3.Peru (Perú), Br4 (Brasil), U19949 (Japón), ATK-1 (Japón), H5 (Japón), BCI2-1 (Canadá), Ni1-3.Peru (Perú), MT4 (Francia), RKI4.Peru (Perú), Bl1.Peru (Perú), Pr52 (Marruecos), BO (Argelia), Pr144 (Marruecos), FrGu1 (Guayana Francesa), NM1626.lac (Guayana Francesa), D13784 (Caribe), pyg19 (África central), ITIS (República Democrática del Congo), PH236 (Gabón), MEL5 (Islas Salomón).
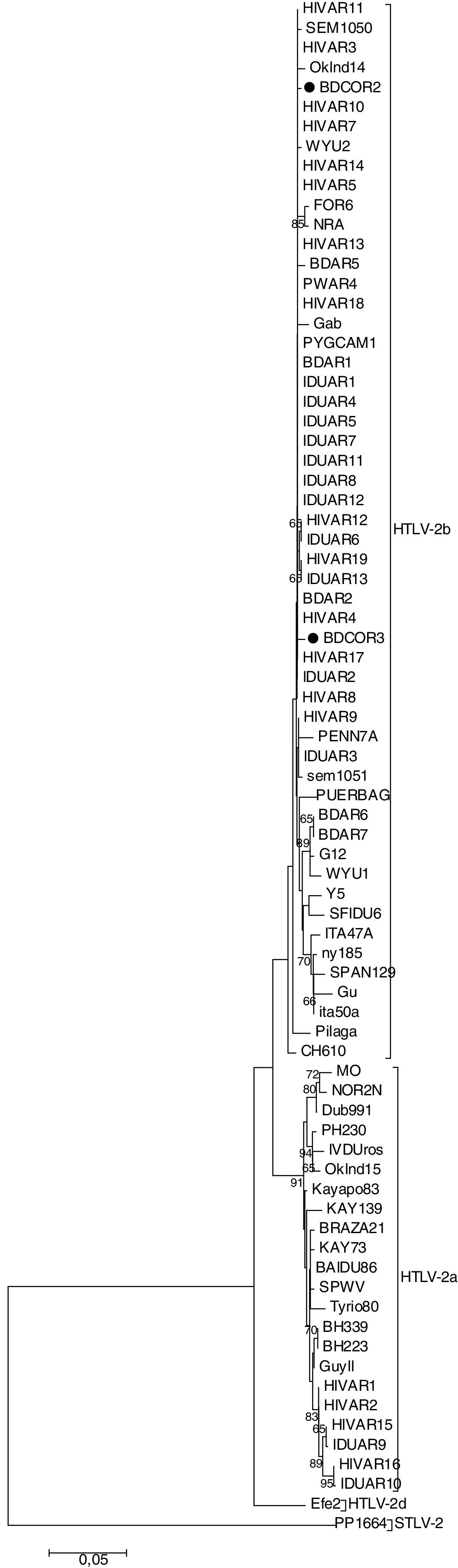
Los 2aislamientos HTLV-2 (BDCOR2 y BDCOR3) se clasificaron dentro del subtipo b junto con secuencias de nuestro país, de Colombia, de EE.UU. y de individuos africanos (fig. 2). BDCOR2 resultó cercanamente relacionada con cepas provenientes de originarios de Argentina (pilagás, FOR6), Gran Chaco (Argentina-Paraguay) (CH610), EE.UU. (SEM1051, PUERBAG), además de donantes de sangre de Buenos Aires (BDAR2) y usuarios de drogas de Argentina (IDUAR3) y de EE.UU. (NRA, Oklnd14, PENN7A). Por otro lado, BDCOR3 se agrupó con secuencias de comunidades originarias de EE.UU. (SEM1050, WYU2), además de cepas reportadas previamente en nuestro país provenientes de individuos infectados con HIV-1 (HIVAR3, HIVAR4, HIVAR5), de un donante de sangre de Formosa (BDAR1) y de uno de la Ciudad de Buenos Aires (BDAR5), de usuarios de drogas residentes en Buenos Aires (IDUAR1, IDUAR2, IDUAR4, IDUAR5, IDUAR6, IDUAR7, IDUAR8, IDUAR11, IDUAR12, IDUAR13) y de una mujer embarazada residente en Buenos Aires (PWAR).
, SEM1050 (EEUU), HIVAR3 (Argentina), Oklnd14 (EEUU), BDCOR2 (Argentina), HIVAR10 (Argentina), HIVAR7 (Argentina), WYU2 (Colombia), HIVAR14 (Argentina), HIVAR5 (Argentina), FOR6 (Argentina), NRA (EEUU), HIVAR13 (Argentina), BDAR5(Argentina), PWAR4 (Argentina), HIVAR18 (Argentina), Gab (Gabón), PYGCAM1 (Camerún), BDAR1(Argentina), IDUAR1 (Argentina), IDUAR4 (Argentina), IDUAR5 (Argentina), IDUAR7 (Argentina), IDUAR11 (Argentina), IDUAR8 (Argentina), IDUAR12 (Argentina), HIVAR12 (Argentina), IDUAR6 (Argentina), HIVAR19 (Argentina), IDUAR13 (Argentina), BDAR2 (Argentina), HIVAR4 (Argentina), BDCOR3 (Argentina), HIVAR17 (Argentina), IDUAR2 (Argentina), HIVAR8 (Argentina), HIVAR9 (Argentina), PENN7A (EE.UU.), IDUAR3 (Argentina), SEM1051 (EE.UU.), PUERBAG (EE.UU.), BDAR6 (Argentina), BDAR7 (Argentina), G12 (XXXX), WYU1 (Colombia), Y5 (Venezuela), SFIDU6 (EE.UU.), ITA47A (Italia), ny185 (EE.UU.), SPAN129 (España), Gu (Europa), ITA50A (Italia), Pilaga (Argentina), CH610 (Paraguay/Argentina), MO (EE.UU.), NOR2N (Europa), Dub991 (Irlanda), PH230 (Camerún), IVDUros (Argentina), Oklnd15 (EE.UU.), Kayapo83 (Brasil), Kay139 (Brasil), BRAZA21 (Brasil), Kay73 (Brasil), SPWV (Brasil), Tyrio80 (Brasil), BH339 (Brasil), BH223 (Brasil), Guyll (Guayana Francesa), HIVAR1 (Argentina), HIVAR2 (Argentina), HIVAR15 (Argentina), IDUAR9 (Argentina), HIVAR16 (Argentina), IDUAR10 (Argentina), Efe2 (Congo), PP1664 (simiana).")
Árbol de maximum likelihood de 78 secuencias de un fragmento de 617pb del LTR del HTLV-2. Las secuencias correspondientes a las muestras analizadas se muestran con ●. La secuencia PP1664 se utilizó como outgroup. Los números en las ramas indican el valor de soporte de cada nodo. El origen geográfico de las secuencias de referencia incluidas en el análisis de arriba hacia abajo son los siguientes: HIVAR11 (Argentina), SEM1050 (EEUU), HIVAR3 (Argentina), Oklnd14 (EEUU), BDCOR2 (Argentina), HIVAR10 (Argentina), HIVAR7 (Argentina), WYU2 (Colombia), HIVAR14 (Argentina), HIVAR5 (Argentina), FOR6 (Argentina), NRA (EEUU), HIVAR13 (Argentina), BDAR5(Argentina), PWAR4 (Argentina), HIVAR18 (Argentina), Gab (Gabón), PYGCAM1 (Camerún), BDAR1(Argentina), IDUAR1 (Argentina), IDUAR4 (Argentina), IDUAR5 (Argentina), IDUAR7 (Argentina), IDUAR11 (Argentina), IDUAR8 (Argentina), IDUAR12 (Argentina), HIVAR12 (Argentina), IDUAR6 (Argentina), HIVAR19 (Argentina), IDUAR13 (Argentina), BDAR2 (Argentina), HIVAR4 (Argentina), BDCOR3 (Argentina), HIVAR17 (Argentina), IDUAR2 (Argentina), HIVAR8 (Argentina), HIVAR9 (Argentina), PENN7A (EE.UU.), IDUAR3 (Argentina), SEM1051 (EE.UU.), PUERBAG (EE.UU.), BDAR6 (Argentina), BDAR7 (Argentina), G12 (XXXX), WYU1 (Colombia), Y5 (Venezuela), SFIDU6 (EE.UU.), ITA47A (Italia), ny185 (EE.UU.), SPAN129 (España), Gu (Europa), ITA50A (Italia), Pilaga (Argentina), CH610 (Paraguay/Argentina), MO (EE.UU.), NOR2N (Europa), Dub991 (Irlanda), PH230 (Camerún), IVDUros (Argentina), Oklnd15 (EE.UU.), Kayapo83 (Brasil), Kay139 (Brasil), BRAZA21 (Brasil), Kay73 (Brasil), SPWV (Brasil), Tyrio80 (Brasil), BH339 (Brasil), BH223 (Brasil), Guyll (Guayana Francesa), HIVAR1 (Argentina), HIVAR2 (Argentina), HIVAR15 (Argentina), IDUAR9 (Argentina), HIVAR16 (Argentina), IDUAR10 (Argentina), Efe2 (Congo), PP1664 (simiana).
El análisis de la región promotora viral (3’LTR) identificó 12 mutaciones en el aislamiento HTLV-1 BDCOR1, al ser comparada con la secuencia prototipo ATK-1, con una identidad nucleotídica del 94,3%. En la tabla 1 se detallan las mutaciones observadas. Además, se realizó una comparación con mutaciones detectadas en secuencias de otras poblaciones de nuestro país, como donantes de sangre, comunidades originarias, trabajadoras sexuales, hombres que tienen sexo con hombres, mujeres embarazadas y pacientes con enfermedades asociadas a la infección por HTLV-1, las cuales se han descripto previamente1,2,5,6,21. Las frecuencias de las mutaciones observadas en las 71 muestras analizadas se detallan en la tabla 2.
Comparación de mutaciones puntuales en la región promotora viral 3’LTR del HTLV-1
| Posición | 8367 | 8403 | 8406 | 8407 | 8408 | 8411 | 8429 | 8479 | 8480 | 8515 | 8562 | 8572 |
| ATK-1 | A | A | - | G | A | G | G | A | A | T | C | G |
| BDCOR1 | C | G | G | A | G | A | A | G | G | C | G | A |
Se comparó la secuencia nucleotídica de un donante de sangre de Corrientes (BDCOR1) con la secuencia de referencia ATK-1.
Frecuencia de mutaciones en 71 secuencias de distintas poblaciones de Argentina
| Frecuencia de mutación% (n) | Cambio nucleotídico respecto de la secuencia prototipo ATK-1 |
|---|---|
| Residentes de Buenos Aires (n = 73) | |
| 100 (71) | G8407A, A8408G, G8411A, G8429A, A8479G, A8480G |
| 97,2 (69) | A8367C |
| 94,4 (67) | G8572A |
| 91,5 (65) | G8515A |
| 90,1 (64) | C8562G |
| 46,5 (33) | A8403G |
| 14,1 (10) | delT8605, C8608T, C8610T |
| 12,7 (9) | T8326C, T8612C |
| 11,3 (8) | C8614T |
| 9,8 (7) | C8606T, T8607C, A8615C |
| 8,5 (6) | A8460G, T8609C |
| 7,0 (5) | G8309A, G8391A, C8624T |
| 5,6 (4) | C8539T, C8562A |
| 4,2 (3) | G8327A, A8338G, A8402G, G8430A, A8456G, G8514A, C8611T, A8615T |
| 2,8 (2) | G8381A, G8435A, A8534G, A8587G, T8607A, C8610A, T8613C,G8619T, G8619A |
| 1,4 (1) | G8322A, C8325T, C8333G, G8351A, C8364G, A8390G, T8401C, A8418G, G8424A, A8434G, G8446A, T8447C, A8455G, A8458G, A8461T, T8525C, T8542C, G8547A, G8552A, C8569T, G8595C, C8608G, C8616A, G8617A, G8617T, C8618T, G8619C, C8620T, G8623C |
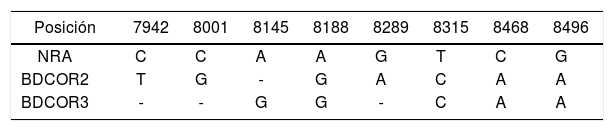
La presencia de mutaciones puntuales en el 3’-LTR de HTLV-2 se determinó por comparación con la secuencia prototipo NRA. El análisis de las secuencias HTLV-2b mostraron una alta similitud tanto entre sí como con la secuencia de referencia NRA, con un valor de 98,8% para BDCOR2 (IC 95%: 97,87-99,74) y de 99,2% para BDCOR3 (IC 95%: 98,05-99,72). Al realizar la comparación, se detectaron 8 mutaciones, de las cuales 4 presentaban cambios nucleotídicos idénticos: A8188G, T8315C, C8468A y C8496A (tabla 3). Cinco de las mutaciones observadas fueron descriptas previamente en secuencias pertenecientes a individuos de diferentes poblaciones de nuestro país (donantes de sangre, embarazadas, comunidades originarias y pacientes con HIV-1)1,2,5,6. Las frecuencias de las mutaciones observadas en las 23 muestras analizadas se detallan en la tabla 4.
Comparación de mutaciones puntuales en la región promotora viral 3’-LTR del HTLV-2b
| Posición | 7942 | 8001 | 8145 | 8188 | 8289 | 8315 | 8468 | 8496 |
|---|---|---|---|---|---|---|---|---|
| NRA | C | C | A | A | G | T | C | G |
| BDCOR2 | T | G | - | G | A | C | A | A |
| BDCOR3 | - | - | G | G | - | C | A | A |
Se compararon las secuencias nucleotídicas de 2 donantes de sangre de Corrientes (BDCOR2 y BDCOR3) con la secuencia de referencia NRA.
Frecuencia de mutaciones en 23 secuencias de distintas poblaciones de Argentina
| Frecuencia de mutación%(n) | Cambio nucleotídico respecto de la secuencia prototipo NRA |
|---|---|
| 100 (23) | G8496A |
| 95,7 (22) | A8188G, T8315C |
| 91,3 (21) | C8468A |
| 17,4 (4) | C7942T |
| 4,3 (1) | A7978C, G7985A, C7994T, C8001G, A8020T, G8056A, G8117A, A8139T, A8145G, T8156G, C8157G, T8158C, T8158G, G8185A, T8250C, G8251A, G8289A, C8336T, T8349C, C8404G, T8440C, G8465A, G8530A |
En Argentina, como ocurre en el resto de Sudamérica, existe una restricción étnico-geográfica para la infección, que circunscribe al HTLV-1 a la familia aymará (kollas) de las tierras altas precordilleranas de Jujuy y Salta y al HTLV-2 a la familia mataco-guaycurú de las zonas bajas de Formosa y Chaco9. En donantes de sangre, la prevalencia de HTLV-1/2 varía desde un 0,03% en áreas no endémicas, como la región central y sur del país, hasta cifras de 0,71% en Salta y 0,96% en Jujuy, en el noroeste5. Con respecto a Corrientes, la prevalencia final confirmada por WB fue de 0,032%, con cifras de prevalencia para ambas infecciones similares a las observadas en áreas no endémicas, incluyendo las descriptas para esta población en la provincia de Misiones y Entre Ríos16,18.
En originarios mbya-guaraníes de Misiones también se reportaron prevalencias muy bajas de HTLV-1/27. Estos datos sustentarían que el área mesopotámica de nuestro país no es una región naturalmente endémica de estos retrovirus.
En cuanto a las características filogenéticas y moleculares de la cepas HTLV-1 y 2, el análisis de la región 3’LTR demostró que la cepa HTLV-1 detectada pertenece al subtipo Cosmopolita, subgrupo A Transcontinental. La secuencia se clasificó dentro del grupo mayoritario latinoamericano, junto a algunas cepas de amerindios del noroeste del país.
Existen reportes previos que describen la circulación de este sugbrupo, tanto en comunidades originarias como en donantes de sangre de áreas endémicas del noroeste, así como también en donantes de sangre, mujeres embarazadas e individuos de poblaciones de riesgo de áreas no endémicas de Argentina1,2,5. Por otro lado, las secuencias analizadas se agruparon con referencias de Brasil y Perú, donde también se ha descripto a este subgrupo, al igual que en el resto del mundo. Por tal razón, existe la posibilidad de que haya existido transmisión (ya sea al sujeto donante directamente, ya sea a individuos con él relacionados —pareja sexual, madre, etc.—) a partir de individuos infectados provenientes de esos países.
En esta investigación no se contaba con datos sociodemográficos de los familiares de los donantes. Y si bien con los datos obtenidos del presente estudio, la provincia de Corrientes puede ser considerada como un área no endémica para HTLV-2, es preciso considerar que existe una afluencia permanente de individuos provenientes de las 2provincias vecinas de Chaco y Formosa, donde el subtipo HTLV-2b ha sido descripto como endémico3,8.
Además, el creciente flujo turístico de los últimos años entre el noreste argentino y Brasil (especialmente el sur de ese país), como también en relación con otras regiones del mundo, incluyendo EE.UU. y Europa —donde el subtipo HTLV-2b está presente, mayoritariamente, en usuarios de drogas inyectables e individuos HIV positivos—, podría estar contribuyendo a la introducción de HTLV-2b en esta provincia15,23.
Sobre la base de los datos epidemiológicos y filogenéticos expuestos, podríamos proponer que, en los 3casos, los virus se han transmitido de madre a hijo, hipótesis que, para ser comprobada, exigiría contar con muestras de las madres, con el fin de demostrar su infección y analizar las secuencias genéticas virales. Además, el hecho de que ninguno de los donantes HTLV-1/2 positivos haya presentado coinfección con HIV, con el virus de la hepatitis B ni con el de la hepatitis C sería una prueba de su falta de antecedentes de drogadicción intravenosa. Sin embargo, no podemos descartar la posibilidad de algún comportamiento de riesgo (uso de drogas inyectables o relaciones sexuales con individuos seropositivos), ya sea de los mismos donantes o de sus parejas sexuales.
Si bien en este estudio se demuestra una prevalencia baja de infección por HTLV-1/2 en donantes de sangre de la provincia de Corrientes, ambos virus se encuentran circulando en individuos sin un antecedente de riesgo parenteral, por lo cual se transforman en relevantes todas aquellas medidas de vigilancia tendientes a disminuir la transmisión sexual y vertical en la población.
Nuestros datos sustentan la importancia de considerar la inclusión del HTLV en un programa de salud de alcance nacional, que permita difundir conocimientos sobre la situación epidemiológica de estos retrovirus y destacar el papel clave de los profesionales de la salud en el diagnóstico. Asimismo, de esa manera se podrán brindar de manera eficiente conocimientos sobre prevención y asistencia médica del individuo infectado por HTLV-1/2.
FinanciaciónEntidad financiadora: Universidad Nacional del Nordeste, Corrientes, Argentina. PI N° 12F018.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Los autores agradecen al Instituto de Cardiología de Corrientes Juana F. Cabral, a la Facultad de Ciencias Exactas y Naturales de la Universidad Nacional del Nordeste y al INBIRS por el apoyo brindado para la publicación de este trabajo.
