


El feocromocitoma es un tumor de baja prevalencia que se origina en las células cromafines de la médula de las glándulas suprarrenales. Estos tumores como el tejido simpático normal se originan del neuroectodermo.
La tríada clásica de presentación clínica es: hipertensión que puede ser persistente, paroxística o fluctuante, cefalea grave pulsátil acompañada de náuseas y/o vómito y palpitaciones con taquicardia o bradicardia refleja; pero también se pueden presentar un gran número de síntomas debido al exceso de catecolaminas plasmáticas, llegando incluso a producir un síndrome coronario agudo.
Su diagnóstico se realiza por medio de la clínica (para la cual se requiere un alto grado de sospecha), el laboratorio y la imagenología. El tratamiento de elección es la resección quirúrgica del tumor por laparoscopia o cirugía abierta.
A phaeochromocytoma is a tumour of low prevalence that originates in the chromaffin cells of the medulla of the adrenal glands. These tumours, like normal sympathetic tissue, originate from neuroectoderm.
The classic triad of clinical findings are: hypertension that can be persistent, paroxysmal or fluctuating, severe throbbing headache accompanied by nausea and/or vomiting, and palpitations with tachycardia or reflex bradycardia. It can also present with a myriad of symptoms due to the excess of plasma catecholamines, even producing an acute coronary syndrome.
Diagnosis is made through the clinical (for which requires a high degree of suspicion), laboratory, and imaging findings. The treatment is surgical resection of the tumour by laparoscopy or open surgery.
Las células cromafines son las encargadas del almacenamiento y producción de las catecolaminas1–6. Los tumores originados en estas células son raros, menos del 1% a nivel mundial7–9 y de acuerdo a su localización son conocidos como: feocromocitoma los que se localizan a nivel de la médula adrenal, representando el 85% de los casos y paragangliomas, aquellos que se ubican a nivel extraadrenal los cuales conforman el 15% restante4,5,9–11.
Anteriormente, se creía que los factores genéticos estaban implicados en el 10% de los feocromocitomas, pero una información reciente sugiere que hasta en el 25% de los casos se pueden detectar mutaciones de la línea germinal. Las causas más frecuentes de predisposición genética son: el Von Hippel Lindau, la neoplasia endocrina múltiple tipo 2, la neurofibromatosis tipo 1 y el síndrome feocromocitoma-paraganglioma7–9,12–14. Los lugares más comunes de presentación extraadrenal son: el órgano de Zuckerkandl (bifurcación de la aorta), la vejiga (< 1%), el mediastino (< 2%), el cuello (< 1%) y otros como el oído medio, el cordón espermático y la base del cerebro4,8,11. Estos tumores pueden encontrarse a cualquier edad pero su mayor incidencia está de los cuarenta a cincuenta años de edad vida7,11.
La mayoría de los feocromocitomas son de carácter benigno y solo un 10% son malignos. Se ubican predominantemente en la glándula suprarrenal derecha siendo estos los de mayores manifestaciones clínicas y cambios en el electrocardiograma1. Es importante tener en cuenta que estos no pueden ser diferenciados por medio de criterios histológicos o bioquímicos y son diagnosticados solo por demostración del tejido tumoral en localizaciones donde comúnmente no existen células cromafines como en el hueso, el hígado, el pulmón y los ganglios linfáticos11.
A continuación se reporta el caso de una paciente atendida en la Clínica Cardio VID por un infarto agudo de miocardio sin elevación del segmento ST secundario a un feocromocitoma y se realiza una revisión de la literatura.
Reporte de casoMujer de 56 años de edad, con antecedente de hipertensión arterial y dislipidemia quien consulta al servicio de urgencias por cuadro clínico de una hora de evolución, consistente en dolor precordial tipo opresivo asociado a mareo, cefalea, náuseas y diaforesis. Al examen físico se encontró pálida con cianosis peribucal, hipertensa 200/90mmHg y saturando 80% al aire ambiente.
Debido al dolor precordial se le realiza un electrocardiograma en el que se observa una bradicardia sinusal, complejos ventriculares prematuros e infradesnivel del segmento ST en la cara anterolateral y pruebas de laboratorio en las que se encontraron: troponinas positivas 10,2 (0,012-0,034), hiperglucemia y leucocitosis.
Se trasladó a la unidad de cuidados intensivos donde ingresó con un edema agudo de pulmón; índice cardiaco de 1,9 L/min/mt2 requiriendo soporte con norepinefrina, dobutamina, milrinone y ventilación mecánica. Allí se le realizó una ecocardiografía transtorácica en la cual se observó un estrés sistólico final de 112 gramos/metro2, un gasto cardiaco sistémico de 2,56/minuto y un ventrículo izquierdo con dilatación leve en sístole de 3,8 centímetros, un estrés sistólico final de 112 gramos/metro2, volumen latido de 25 mililitros y un gasto cardiaco sistémico de 2,56/minuto.
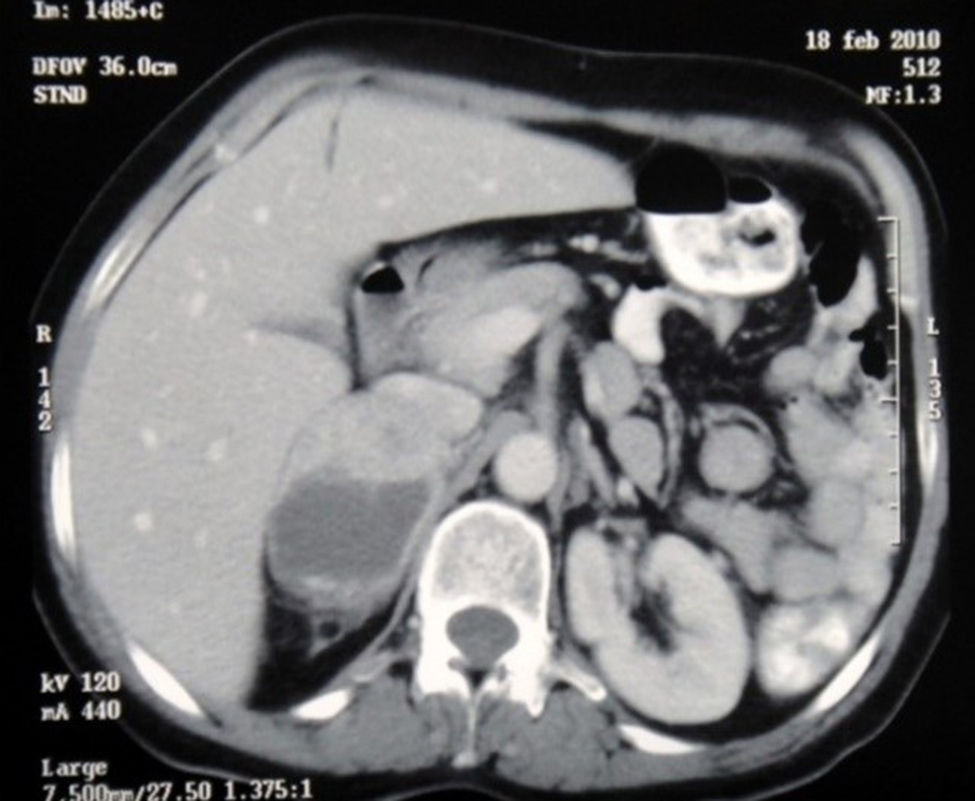
Durante su estancia en la unidad de cuidados intensivos presentó fiebre, elevación de los reactantes de fase aguda y cifras tensionales con tendencia a la hipotensión, sin evidencia de foco por lo que se inició manejo antibiótico empírico con meropenem. Se solicitaron paraclínicos e imagenología para buscar el foco infeccioso, dentro de ellos una tomografía axial computarizada toracoabdominal, en la cual se encontró consolidación de ambas bases pulmonares y de manera llamativa, una masa ubicada sobre el riñón derecho que se extendía al hígado (fig. 1). En el aspirado traqueal se aisló un Staphylococcus aureus sensible a meticilina y una Klebsiella pneumoniae por lo que se descartó manejo antibiótico a piperacilina tazobactam con lo que presentó resolución de su cuadro respiratorio y a los 23 días se extubó sin complicaciones.

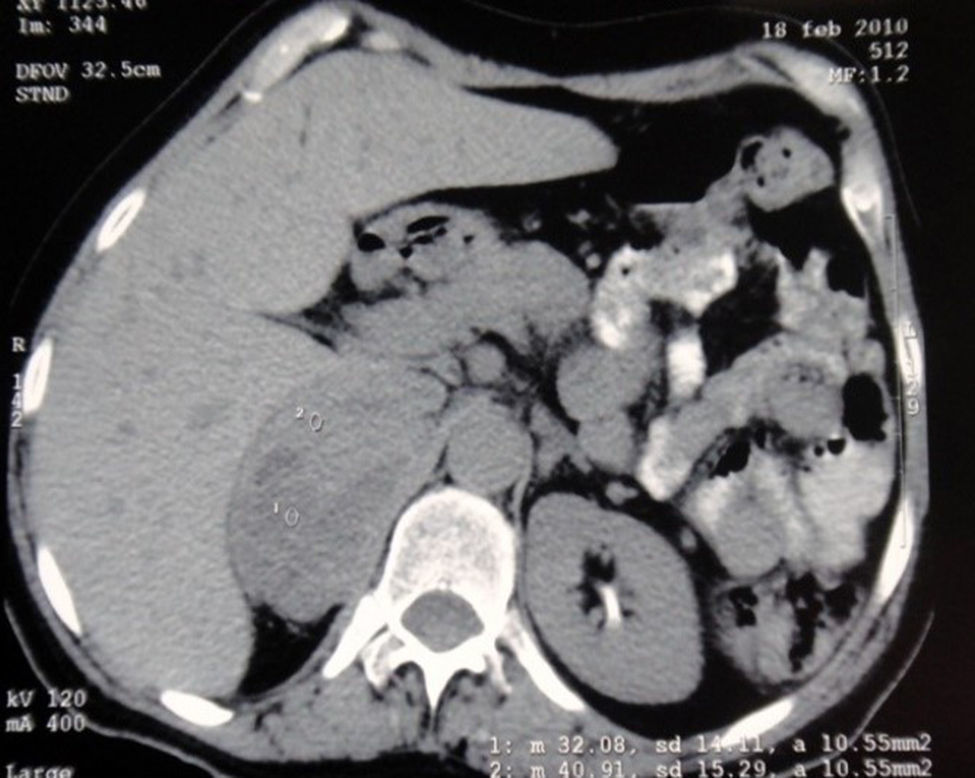
Después de una estancia de veinticuatro días en la unidad de cuidados intensivos fue traslada a hospitalización, donde se solicitaron estudios del incidentaloma adrenal, las cuales fueron positivas: metanefrinas en orina 6,451 ug/24 horas (0,0 -1000), adrenalina y noradrenalina en orina 139,8 ug/24 horas (< 20 ug/24 horas) y 135,6 ug/24 horas (10-70 ug/24 horas), respectivamente. Se inició manejo con prazosina, metoprolol y posteriormente se realizó supraadrenelectomía con resección de masa de 8×8 centímetros ubicada en la glándula suprarrenal derecha (fig. 2).

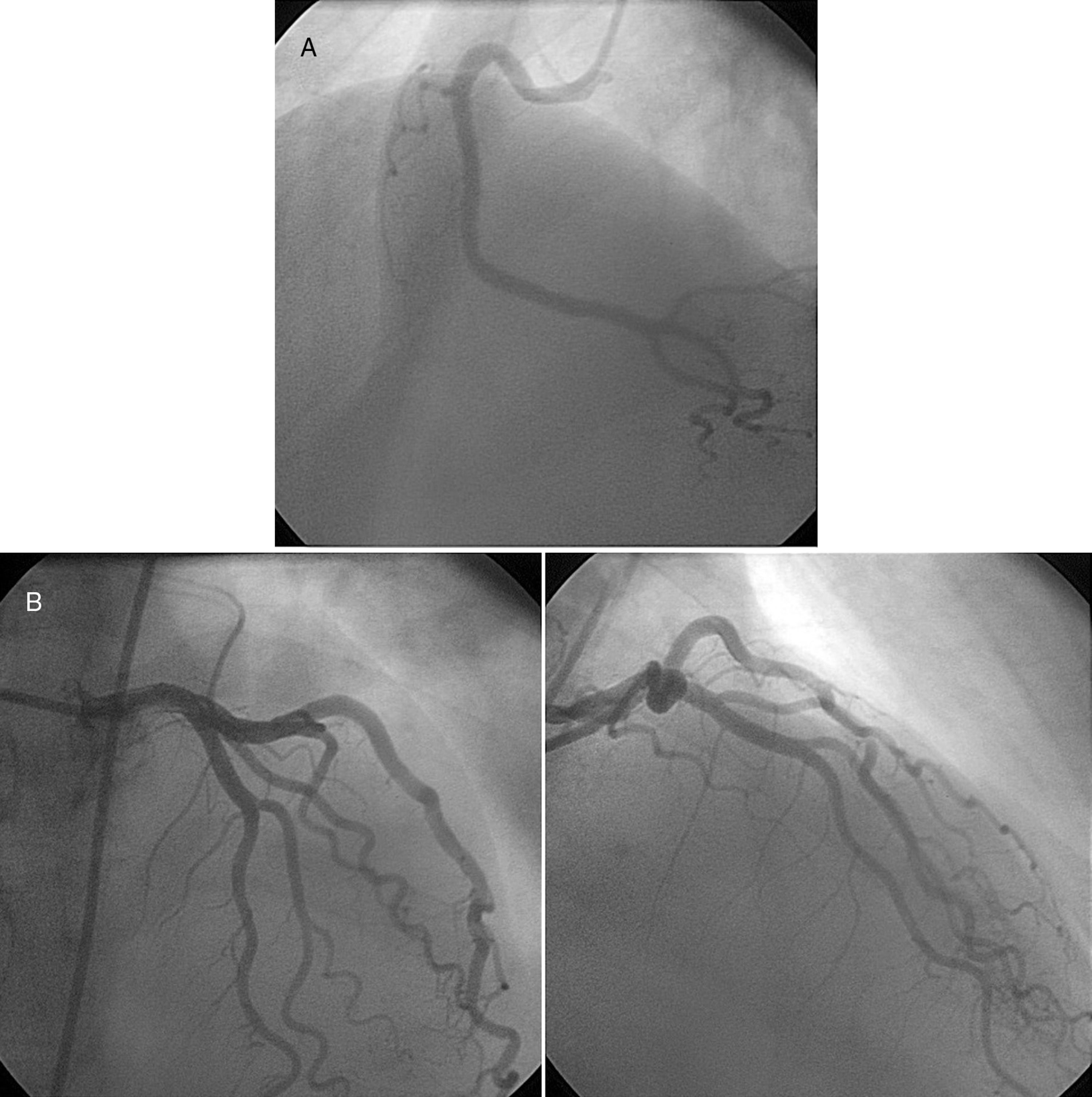
Ocho días después de la cirugía es llevada a coronariografía (fig. 3), en la cual se encontró: el tronco principal izquierdo sin lesiones obstructivas, la arteria descendente anterior sin lesiones obstructivas visibles, la arteria circunfleja sin lesiones obstructivas, la coronaria derecha dominante sin lesiones ateromatosas obstructivas y la D1 con trayecto fistuloso al ventrículo izquierdo. Con este resultado se descartó la enfermedad coronaria como causa del infarto agudo de miocardio.
 arteria coronaria derecha. B) arteria coronaria izquierda.")
El estudio ecocardiográfico de control realizado un año después fue normal, sin trastornos segmentarios de la contracción y con fracción de eyección del ventrículo izquierdo del 60%.
Actualmente la paciente no se encuentra recibiendo medicamentos antihipertensivos, solo inhibidores de la enzima convertidora de angiotensina a bajas dosis para evitar la remodelación ventricular.
DiscusiónEn las glándulas suprarrenales se pueden encontrar los incidentalomas, los cuales hacen parte de las «enfermedades de la tecnología moderna», pues son tumores cuya existencia es desconocida antes del estudio imagenológico y se encuentran accidentalmente cuando este se está realizando. No se debe clasificar como incidentaloma adrenal aquel tumor que se encuentra durante una búsqueda de metástasis o cuando hay sospecha previa de anormalidades adrenales.
Los incidentalomas adrenales pueden ser: no secretores, hipersecretores, carcinomas adrenales primarios u otras masas adrenales y metástasis. Dentro de los tumores hipersecretores se encuentra el feocromocitoma12,15, cuya tríada clásica es: la hipertensión arterial que puede ser persistente, paroxística o fluctuante, la cefalea grave pulsátil acompañada de náuseas y/o vómito y las palpitaciones con taquicardia o bradicardia refleja3,15. En la mayoría de los pacientes con feocromocitoma no se encuentra esta tríada y en pacientes con la hipertensión esencial se pueden presentar síntomas paroxísticos7. La hipertensión paroxística o sostenida es el signo más común del feocromocitoma, pero el 5-15% de los pacientes pueden ser normotensos. La frecuencia de normotensión es mayor en pacientes con incidentalomas adrenales o en aquellos en los que se busca el feocromocitoma familiar. La cefalea puede ser leve o grave y de duración variable, esta ocurre en el 90% de los pacientes sintomáticos.
Otros síntomas que pueden estar presentes son: la sudoración generalizada, los cambios en el peso con tendencia a la pérdida por el hipermetabolismo, la hipotensión (principalmente en tumores donde predomina la secreción de epinefrina), el temblor fino, la hematuria no dolorosa, el síncope, la disnea, el fenómeno de Raynaud, la visión borrosa e incluso el compromiso cardiaco1,2,4,11.
El espectro cardiaco puede ir desde una arritmia supraventricular o ventricular hasta una miocardiopatía grave o choque cardiogénico, debido a los altos niveles de catecolaminas séricas que provocan la vasoconstricción y el aumento de la demanda de oxígeno, produciendo así una necrosis por isquemia1,2.
El diagnóstico del feocromocitoma se realiza por medio de la clínica, el laboratorio y la imagenología. En el laboratorio lo más utilizado son las catecolaminas y metanefrinas en plasma y las metanefrinas fraccionadas en orina de 24 horas. Si estas pruebas son positivas se debe proceder a realizar los estudios imagenológicos, como son la tomografía computarizada contrastada de abdomen, la resonancia magnética y la gammagrafía adrenal. La tomografía computarizada de abdomen es la más utilizada por ser la más costo-efectiva1,2,8,10–13,15.
El tratamiento de elección es quirúrgico ya sea por laparoscopia o por cirugía abierta. Antes de la cirugía se debe normalizar la presión arterial, debido a que la incidencia de complicaciones perioperatorias se encuentran vinculadas con la presión arterial preoperatoria.
El control de la presión arterial requiere alfa y beta antagonistas, comúnmente se utilizan los alfa-1 antagonistas como la prazosina o la doxazosina porque tienen un tiempo de acción corto y no producen taquicardia mediada por el bloqueo de los receptores alfa 2. La primera dosis de prazosina puede inducir una caída brusca de la presión arterial, por ello su dosis debe ir aumentando progresivamente desde 0,5 a 5mg tres veces al día. El bloqueo alfaadrenérgico, generalmente, produce taquicardia debido a la estimulación del beta-receptor por las catecolaminas, por lo que se debe adicionar el beta-bloqueador2,4,11.
Para confirmar la cura del tumor se deben realizar pruebas bioquímicas, pero no inmediatamente, debido a que durante la primera semana posquirúrgica se pueden seguir secretando catecolaminas. Deben realizarse aproximadamente el día 10 después de la resección del tumor. Si la concentración de metanefrinas continúa elevada se debe realizar una gammagrafía adrenal. Debido a que los feocromocitomas o paragangliomas pueden ser recurrentes, se deben hacer anualmente controles de la presión arterial y de los niveles de metanefrinas4,8,9,12.
La paciente presentó un infarto agudo de miocardio sin elevación del ST con una coronariografía sin alteraciones, por lo que se concluyó que el incidentaloma adrenal derecho fue el causante de este.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflictos de interesesLos autores declaran no tener ningún conflicto de intereses.