




El síndrome de la plaqueta pegajosa en un trastorno cualitativo plaquetario en el que bajas concentraciones de epinefrina y adenosina difosfato producen hiperagregabilidad plaquetaria considerable. Se ha especulado mucho sobre la etiología de este trastorno sin que sean claros sus mecanismos fisiopatológicos. Desde el punto de vista clínico, se asocia a trombosis arteriales y venosas recurrentes en pacientes jóvenes, pérdidas gestacionales, otras complicaciones obstétricas y cefalea recurrente.En la literatura se ha descrito su presentación familiar, lo que hace sospechar su comportamiento hereditario autosómico dominante; también se ha reportado un fenotipo adquirido de la enfermedad en algunas poblaciones especiales como pacientes con enfermedad renal crónica en terapia de reemplazo renal o posterior al trasplante renal y en pacientes con cuadros inflamatorios o inmunosupresión. Se expone el caso de una paciente con antecedente de cefalea de difícil manejo, síndrome hipertensivo asociado al embarazo y mortinato, con síndrome del nodo enfermo y disautonomía manejadas con implantación de marcapasos definitivo bicameral con sensor CLS, que desarrolló trombosis de la vena subclavia, asociada al electrodo de marcapasos, recurrente a pesar de anticoagulación con warfarina y rivaroxabán e incluso a pesar de antiagregación con ácido acetilsalicílico, con posterior diagnóstico de síndrome de la plaqueta pegajosa.
Sticky platelet syndrome is a qualitative platelet disorder in which low concentrations of adrenaline and adenosine diphosphate produce considerable platelet hyperaggregability. There has been much speculation on the origin of this disorder as its pathophysiological mechanisms of action are not yet clear. From a clinical point of view, it is associated with recurrent arterial and venous thrombosis in young patients, miscarriages, other obstetric complications and recurrent headaches.Its familial presentation has been described in the literature, suspecting that it is of a dominant autosomal hereditary nature. An acquired phenotype of the disease has also been reported in some particular patients, such as patients with chronic kidney disease on renal replacement therapy or after a kidney transplant, as well as in patients with inflammatory processes or immunosuppression. The case is presented of a patient with a history of difficult to manage headaches, a hypertensive syndrome associated with the pregnancy, and a foetal death. She also had sick sinus node syndrome and dysautonomia managed with a definitive dual-chamber pacemaker with a CLS sensor. There was then a thrombosis in the subclavian vein associated with the pacemaker electrode, being recurrent despite anticoagulation with warfarin and rivaroxaban, and even despite anti-aggregation treatment with acetyl salicylic acid. She was subsequently diagnosed with sticky platelet syndrome.
Hasta el momento han sido bien estudiados y descritos en la literatura los defectos en los factores de la coagulación de los que se derivan los estados de hipercoagulabilidad y sus manifestaciones trombóticas; sin embargo, hay menor entendimiento de los desórdenes cualitativos y cuantitativos de las plaquetas que favorecen los estados protrombóticos; un ejemplo de ello es la reciente descripción del defecto Wien-Penzing en 19911. A la fecha, existen 59 publicaciones en la base de datos Pubmed sobre el síndrome de la plaqueta pegajosa. Esta es una entidad caracterizada por hiperagregabilidad plaquetaria inducida por bajas cantidades de adenosín difosfato y/o epinefrina, tanto o más prevalente que otras trombofilias como el síndrome antifosfolípido, asociada a manifestaciones trombóticas arteriales y venosas recurrentes y a pérdidas gestacionales tempranas. Su descripción reciente y el desconocimiento de la entidad contribuyen al subdiagnóstico de la misma.
CasoPaciente de 36 años con antecedente de cefalea migrañosa de difícil manejo, con embarazo previo en el que presentó síndrome hipertensivo asociado y mortinato, que consultó con cuadro clínico de varios años de evolución, conformado por mareos y síncopes recurrentes y prueba de mesa basculante positiva para síncope mixto. Se inició manejo con cambios del estilo de vida y fludrocortisona, sin mejoría significativa.
Durante el seguimiento persistió con sintomatología vasovagal, fatiga, limitación de su clase funcional, hipotensión y bradicardia, por lo que se realizó prueba ergométrica y monitorización Holter, en la que se evidenció disfunción sinusal. Se descartaron alteraciones metabólicas (carbohidratos y lípidos), tiroideas e hidroelectrolíticas. Por lo anterior, se llevó a estudio electrofisiológico que demostró disfunción sinusal y se decidió implante de marcapasos con sensor CLS para su síndrome del nodo enfermo y su disautonomía.
Tres meses después, presentó edema importante en miembro superior izquierdo, documentándose trombosis venosa profunda de la vena subclavia, por lo que se realizó angioplastia con balón e implantación de stent. Se inició anticoagulación con warfarina. Dos meses más tarde presentó nuevamente edema y dolor en la extremidad superior izquierda; en venografía se encontró trombosis intrastent y se realizó angioplastia e implante de stent intrastent. Continuó anticoagulación con warfarina con INR dentro de metas pero con múltiples manifestaciones hemorrágicas espontáneas como gingivorragia o hematoma espontáneo en muslo derecho. Dos meses después de la implantación del segundo stent consultó con cuadro clínico consistente en trombosis venosa profunda aguda de la extremidad superior izquierda por lo que fue llevada a nueva venografía con evidencia de trombo intrastent no oclusivo, luego de lo cual se decidió cambio de tratamiento médico con inicio de anticoagulación con rivaroxabán 20mg/día.
Consultó de manera recurrente por cefaleas que ameritaron interconsulta con Neurología; fue difícil el control del dolor y no se documentaron lesiones intracraneales ocupantes de espacio o complicaciones hemorrágicas asociadas. Un año después presentó recurrencia del cuadro y en nuevo procedimiento se evidenció oclusión del stent por trombo extensivo a venas axilar y humeral izquierda, con circulación por colaterales desde tronco venoso tirocervical hacia yugular interna e innominada. Fue llevada a trombólisis selectiva con rTPA y se realizó angioplastia con balón liberador de paclitaxel. Se decidió adicionalmente antiagregacióón con ácido acetil salicílico 100mg/día. Para este entonces se habían realizado estudios seriados de hipercoagulabilidad con los cuales se descartaron trombofilias adquiridas y congénitas como deficiencia de las proteínas C y S, resistencia a la proteína C activada, mutación del factor V de la coagulación (factor V de Leiden), mutación de la antitrombina 3, síndrome antifosfolípido, síndrome nefrótico, neoplasia activa de base e hiperhomocisteinemia, entre otros.
Unos meses después tuvo recurrencia del dolor y edema en la extremidad superior izquierda. Se complementaron estudios con ecocardiografía transtorácica que descartó trastornos segmentarios de la movilidad o disfunción sistólica sospechosa de cardiopatía isquémica. Una vez más se hizo venografía donde se encontró trombosis intrastent pero abundante circulación colateral con drenaje eficiente, por lo cual se decidió no realizar nuevamente intervencionismo.
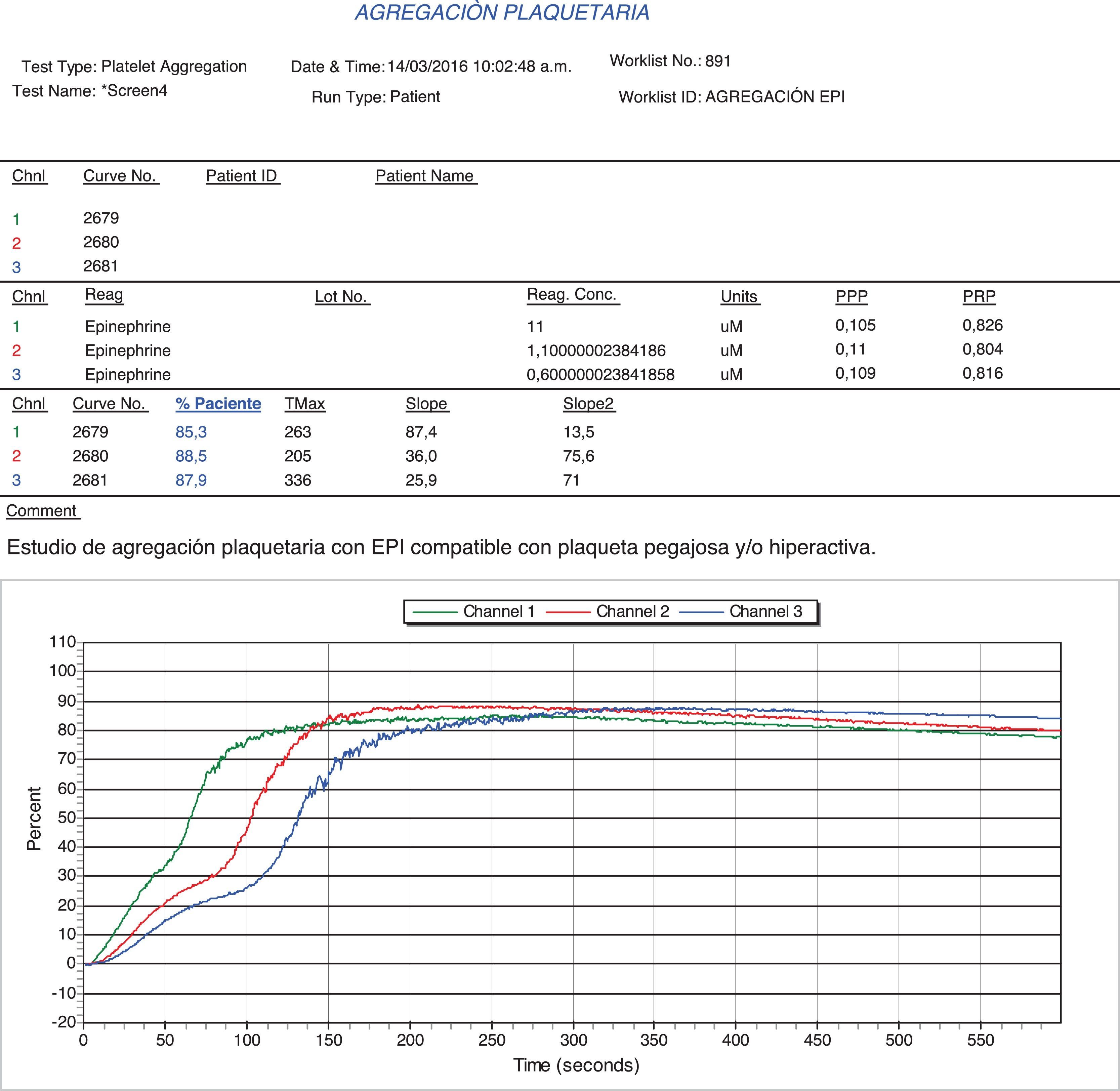
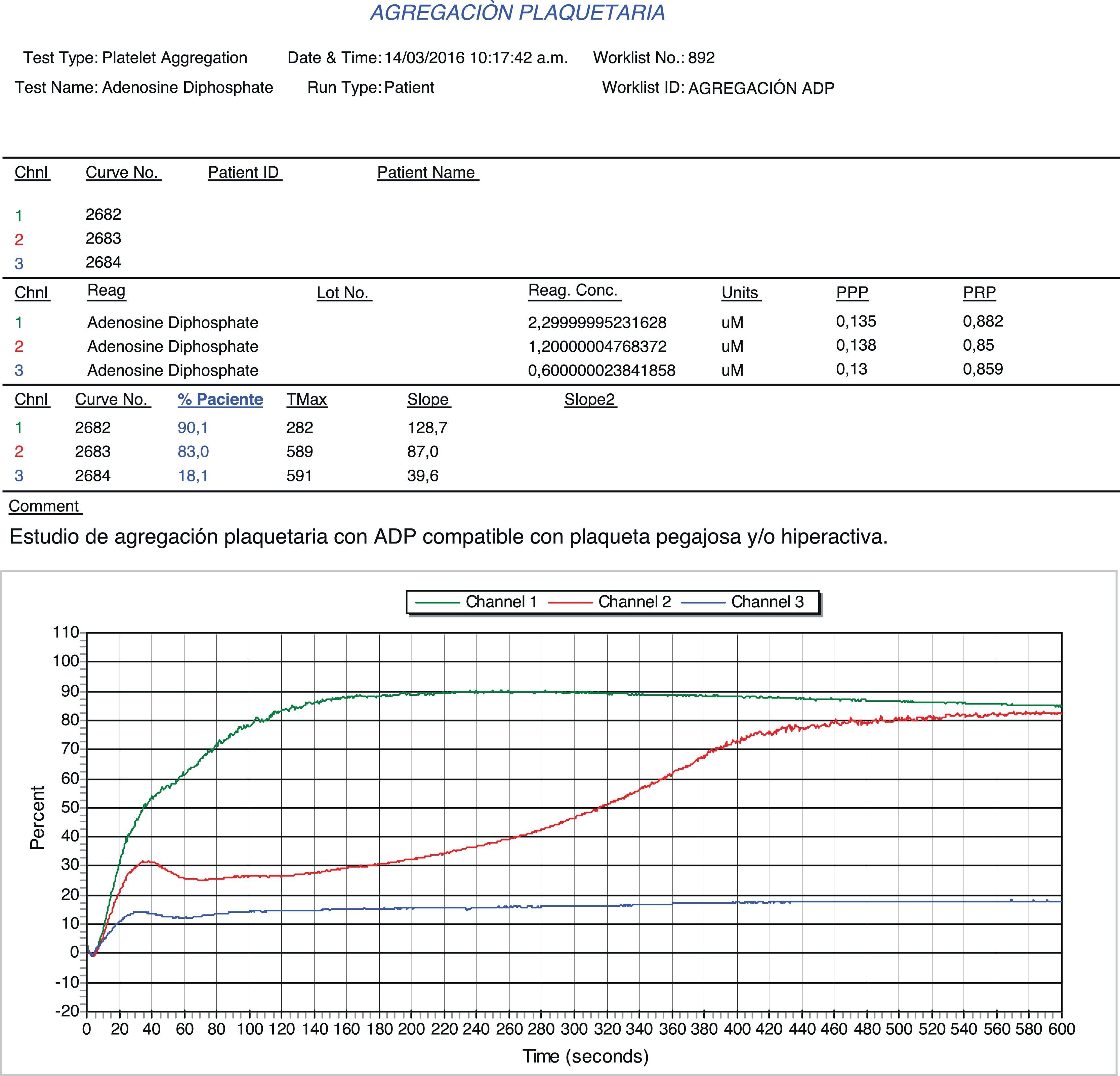
La paciente fue valorada de manera conjunta por los servicios de Hemodinamia, Electrofisiología y Hematología. Se realizaron estudios de extensión incluida agregometría plaquetaria que demostró hiperagregabilidad con epinefrina y con ADP (figs. 1 y 2), por lo que se decidió continuar tratamiento con rivaroxabán y clopidogrel teniendo en cuenta la recurrencia de los fenómenos trombóticos a pesar de la terapia dual (anticoagulación con inhibidor directo del factor Xa y antiagregación con ASA). Se decidió continuar monoterapia con clopidogrel teniendo en cuenta la refractariedad al tratamiento con ASA y anticoagulación. Desde entonces permanece en seguimiento clínico y no se han documentado nuevos episodios de trombosis de la vena subclavia o en otro territorio vascular.
Historia del síndrome de la plaqueta pegajosa
.")
Las plaquetas pueden presentar desórdenes cualitativos y cuantitativos que, a su vez, pueden ser de naturaleza hemorrágica o trombótica. Fueron Bilzzozero y Osler en 1873, quienes hicieron la primera descripción de la plaqueta como componente celular de la sangre y su asociación con la coagulación. Posteriormente se documentó su presencia en los trombos de placas ateroscleróticas, vegetaciones valvulares y aneurismas aórticos. Hacia 1910, Duke describió la relación inversamente proporcional entre el recuento plaquetario y el sangrado2.
Desórdenes plaquetarios cualitativos hemorrágicos como la anomalía de May-Hegglin, la trombastenia de Glanzmann y el síndrome de Bernard-Soulier fueron ampliamente descritos a inicios del siglo xx, mientras que los desórdenes plaquetarios cualitativos trombóticos solo empezaron a describirse en 1979 cuando Al Mefty publicó una serie de 22 pacientes con isquemia cerebral transitoria sin enfermedad aterosclerótica por angiografía, lesiones intracraneanas por TAC, evidencia de cardioembolia, arteritis, colagenopatía o antecedente de migraña, en quienes el único hallazgo común fue el aumento de la agregabilidad plaquetaria. Describió también el efecto terapéutico de la Aspirina y la recurrencia de los eventos trombóticos con su descontinuación2,3.
En 1983, Holidey estableció la relación entre la epinefrina y el ADP con la hiperagregabilidad, y reportó su asociación con trombosis arteriales en pacientes jóvenes.
La definición inicial del síndrome de la plaqueta pegajosa fue planteada por Mammen en 1984, como un trastorno autosómico dominante caracterizado por hiperagregabilidad plaquetaria in vitro como respuesta a bajas concentraciones de ADP y/o epinefrina, en la que no hay respuesta a otros inductores como el colágeno, ácido araquidónico, ristocetina o trombina1,4,5.
En 1986 Rubenfire documentó el trastorno en 41 pacientes con síndrome coronario agudo3, y en 1995 nuevamente Mammen estableció la relación del trastorno con complicaciones gestacionales y la presentación familiar del mismo. En su serie de más de 200 pacientes jóvenes reportó tanto trombosis arteriales como venosas, y postuló la hipótesis de que el estrés fisiológico podía desencadenar eventos trombóticos. Describió los subtipos 1 y 2 de acuerdo con la respuesta a ADP y epinefrina en la prueba de agregometría4.
Fue en 1998 cuando Brick describió el subtipo 3, el más infrecuente de todos, donde el trastorno de la agregación aparece en respuesta a ADP de manera aislada. Brick reportó además que hasta un 20% de los abortos recurrentes podrían ser explicados por el síndrome de la plaqueta pegajosa, así como la quinta parte de los fenómenos trombóticos arteriales1.
En los últimos años, Chitoor documentó el fracaso de la anticoagulación en fenómenos tromboembólicos venosos recurrentes y planteó la hipótesis de la multifactorialidad de esta trombofilia y su coexistencia con otras trombofilias. De igual forma, Yagmur reportó la asociación con la enfermedad renal crónica, la hemodiálisis y el trasplante renal6.
Recientemente Lewerenz y Moncada describieron algunas manifestaciones cutáneas asociadas como atrofia, livedo reticularis, eritema nudoso, fenómeno de Raynaud, entre otros2,7. Velásquez-Sánchez-de-Cima reportó el uso potencial del clopidogrel y la antiagregación dual2.
FisiopatologíaLas plaquetas, responsables de la hemostasia primaria, son fragmentos citoplasmáticos pequeños derivados de los megacariocitos con una vida media de 7-10 días. Poseen gránulos densos ricos en adenosín difosfato y calcio, y gránulos alfa ricos en factor de Von Willebrand (cuyo receptor es la glicoproteína Ib) y fibrinógeno (cuyo receptor es la glicoproteína IIb/IIIa). La trombogénesis se inicia como respuesta a la lesión endotelial, en la que hay exposición del colágeno subendotelial que se une el factor de Von Willebrand circulante. La glicoproteína Ib plaquetaria se une al factor de Von Willebrand, con lo cual se inducen cambios conformacionales que resultan en la liberación de adenosín difosfato que, a través de la estimulación de los receptores P2Y12 y P2Y1, activa la plaqueta e induce la liberación de fibrinógeno, la exposición de glicoproteína IIb/IIIa y la salida de calcio para activar más plaquetas y la cascada de la coagulación.
La agregación plaquetaria es mediada por la glicoproteína IIb/IIIa que une cada extremo de una molécula de fibrinógeno circulante de tal manera que dos plaquetas adyacentes puedan agruparse, fenómeno que al magnificarse produce la formación del coágulo. Trastornos cualitativos y cuantitativos de algunos de los mediadores descritos previamente, conducirán a trastornos plaquetarios que se traducen en trombofilias o complicaciones hemorrágicas.
Las trombofilias son condiciones que pueden tener cursos asintomáticos o asociarse a enfermedades tromboembólicas. Cuando se asocian a defectos congénitos se trata de trombofilias primarias o hereditarias y cuando se asocian a enfermedades como el cáncer, la exposición a fármacos como los anticonceptivos orales, a estados fisiológicos como la gestación y el periparto, se tratará entonces de trombofilias secundarias o adquiridas.
El síndrome de la plaqueta pegajosa es una trombocitopatía clásicamente descrita como una trombofilia primaria hereditaria con patrón autosómico dominante, en la que hay un desorden cualitativo de la agregación plaquetaria determinado por hiperagregabilidad en respuesta a epinefrina y adenosín difosfato, que favorece fenómenos trombóticos arteriales y venosos recurrentes, así como pérdidas gestacionales tempranas3. Estudios de núcleos familiares de pacientes “caso índice” avalan el patrón autosómico dominante de herencia8. En la actualidad, la literatura sugiere que es el defecto plaquetario hereditario más común, con frecuencia igual o incluso superior a la del síndrome antifosfolípido.
Algunos trabajos han propuesto hiperagregabilidad plaquetaria inducida por mediadores diferentes a adenosín difosfato y epinefrina, como por ejemplo la hipersensibilidad al colágeno y la sobreexpresión de TRAP-6 (péptido activado por el receptor de trombina 6).
Hasta ahora se atribuye este síndrome a diferentes mutaciones en mediadores de la activación plaquetaria. Mammen describió que mutaciones en diferentes glicoproteínas de membrana de las plaquetas se asocian a aumento de la agregabilidad. Múltiples autores documentan el aumento en la expresión de CD62 – P selectina, CD 63, CD 61 y CD51 (estos dos últimos receptores para la vitronectina) en la superficie de las mismas, que sugiere hiperactividad además de hiperagregabilidad9,10.
Polimorfismos de la GPIIIa PLA A1/A2 parecen tener también alguna asociación, aunque hasta ahora los estudios no son concluyentes11. La proteína GAS6, miembro de las proteínas dependientes de la vitamina K y presente en los gránulos alfaplaquetarios, tiene alta homología estructural con la proteína S de la coagulación, así como alta interacción con las tirosina kinasas que a ella se asocian, al igual que con los receptores plaquetarios para ADP, por lo cual se considera actúa directamente como estimulante de su activación y agregación12. Sin embargo, no todos los estudios han logrado establecer una relación entre el polimorfismo de la proteína GAS6 y el síndrome de la plaqueta pegajosa13.
Así mismo, el receptor endotelial para la agregación plaquetaria 1 PEAR1, con dominio extracelular similar al del factor de crecimiento epitelial y presente en plaquetas y endotelio, es fosforilado con el contacto entre las primeras para favorecer su activación. Polimorfismos en PEAR1 se han asociado con aumento de eventos trombóticos y menor respuesta a la Aspirina12.
La glicoproteína 6 es una proteína transmembrana de la plaqueta que interactúa con el colágeno expuesto en el endotelio lesionado para facilitar su activación y adhesión. Múltiples polimorfismos de la misma encontrados en pacientes con diagnóstico de síndrome de la plaqueta pegajosa se han asociado con abortos recurrentes14–17. Debe anotarse que a pesar de múltiples factores propuestos dentro de la fisiopatología del síndrome de la plaqueta pegajosa, a la fecha no existe claridad sobre los mecanismos causales de este desorden cuya naturaleza es multifactorial18.
Descripciones recientes han propuesto además una forma adquirida del síndrome de la plaqueta pegajosa de manera aislada en casos en los que no se identifica un componente familiar, o también en relación a trastornos metabólicos como la diabetes mellitus y la aterosclerosis, trastornos inflamatorios como la sepsis, pacientes con inmunosupresión farmacológica, pacientes con virus de la inmunodeficiencia humana (VIH) en tratamiento antirretroviral y pacientes con enfermedad renal crónica, en especial aquellos llevados a trasplante renal.
Se propone que el estrés fisiológico y la liberación de catecolaminas incrementa la agregabilidad plaquetaria. En otros fenómenos inflamatorios se considera que el daño endotelial induce adhesión y activación y perpetúa la agregabilidad de las plaquetas.
In vitro se ha observado como la ciclosporina A y el tacrolimus aumentan la agregación plaquetaria a través de la fosforilación de proteínas moduladoras; la ciclosporina además aumenta prostanoides y endotelina, y el tacrolimus y el sirolimus tienen potenciales efectos pronecróticos y antiangiogénicos con los que aumenta el daño endotelial y posiblemente el ATP almacenado a nivel granular, así como su liberación y por ende la activación plaquetaria.
No puede descartarse que este síndrome se trate de una condición genotípica cuya manifestación fenotípica requiere de un segundo estímulo, por ejemplo los fenómenos inflamatorios, de acuerdo con lo referido en múltiples reportes de caso.
ClasificaciónEl síndrome de la plaqueta pegajosa se clasifica de acuerdo con el resultado de la agregometría plaquetaria según respuesta a diferentes concentraciones de los reactantes adenosín difosfato (ADP) y epinefrina (tabla 1).
EpidemiologíaSe desconoce la prevalencia del síndrome de la plaqueta pegajosa. De acuerdo con la literatura disponible este síndrome explicaría el 20 al 28% de las enfermedades trombóticas en general, el 20% de los casos de trombosis arteriales inusuales y el 13% de las trombosis venosas profundas inexplicadas. Adicionalmente, puede contribuir en el 20% de los casos de infarto agudo del miocardio, isquemia cerebral transitoria y ataque cerebrovascular. Como se mencionó anteriormente, diferentes series han descrito el síndrome de la plaqueta pegajosa en hasta el 20% de las pérdidas fetales tempranas3.
En la enfermedad renal crónica se ha descrito una prevalencia del 82% en pacientes en hemodiálisis y del 67% en pacientes con transplante renal6. Se cree que está asociado al 44% de las trombosis recurrentes de accesos vasculares en pacientes en hemodiálisis. También debe sospecharse en pacientes con enfermedad de injerto contra huésped1,19.
Ruiz-Argüellez et al. en su trabajo sobre trombofilias en mestizos mexicanos encontraron que el síndrome de la plaqueta pegajosa puede cursar de manera concomitante con otras anormalidades protrombóticas. Describieron el componente familiar de la misma y la evolución clínica satisfactoria una vez iniciada la antiagregación con ácido acetil salicílico documentada por normalización de la agregometría plaquetaria durante el tratamiento hasta en el 75% de los casos, con necesidad de antiagregación dual con clopidogrel solo en el 25% de los casos restantes y una recurrencia de episodios vasooclusivos del 3,6% en su seguimiento promedio de 13 meses. Encontraron además que hasta el 32% de las mujeres con SPP han presentado un aborto previamente en sus vidas20.
En 2004 se describió en Colombia una serie de casos de mujeres con antecedente de trombosis venosa y pérdidas gestacionales en el hospital Militar Central en Bogotá, en cuyo estudio se documentó el síndrome de la plaqueta pegajosa21.
Posteriormente, en el Laboratorio Clínico Hematológico en Medellín en 130 pacientes a quienes se les ordenó agregometría plaquetaria entre 2013 a 2014 (17 meses) y diagnosticó síndrome de la plaqueta pegajosa, se encontró de manera consecuente con otros trabajos reportados, mayor incidencia en mujeres que en hombres (30% hombres, 70% mujeres), y que el fenotipo más prevalente de la enfermedad en nuestro medio es el tipo 1 (70%) seguido de los tipos 2 y 3 (29,2% y 0,8% respectivamente), como se ha descrito en la literatura mundial3.
Manifestaciones clínicasEl síndrome de la plaqueta pegajosa se asocia con manifestaciones trombóticas en pacientes jóvenes, usualmente menores de 40 años, sin otros factores de riesgo conocidos. Con frecuencia se observan trombosis arteriales pero también venosas en localizaciones inusuales (venas y arterias de la retina, senos cerebrales) y en algunos casos son recurrentes a pesar de anticoagulación óptima, especialmente con warfarina. En muchos de estos casos, es claro el antecedente familiar de manifestaciones trombóticas22.
No es infrecuente en estos pacientes el antecedente de migraña ni en las mujeres el antecedente de insuficiencia placentaria, síndromes hipertensivos asociados al embarazo, restricción del crecimiento intrauterino y abortos previos; así mismo manifestaciones trombóticas precipitadas por anovulatorios y hormonas de sustitución.
Debe considerarse el síndrome de la plaqueta pegajosa en la enfermedad tromboembólica venosa, incluida tromboembolia pulmonar, especialmente en ausencia de factores desencadenantes claros23.
Hasta el 50% de los pacientes presentará trombosis de vasos retinianos. Existen amplios reportes de caso en la literatura sobre complicaciones oftalmológicas diferentes y complejas, incluido el daño del nervio óptico24–26.
También debe descartarse esta condición de base en pacientes con complicaciones trombóticas alrededor de procedimientos invasivos como en el caso reportado de trombosis durante endarterectomía carotídea27.
DiagnósticoEn la agregometría plaquetaria de los pacientes con síndrome de la plaqueta pegajosa, es normal la respuesta a estimulantes como colágeno, ácido araquidónico y ristocetina.
En la prueba de agregabilidad con epinefrina y adenosín difosfato se exponen las plaquetas a diferentes concentraciones de las mismas y se valora la respuesta comparada con parámetros de normalidad para la prueba. Debe recordarse que la función plaquetaria tiene variación circadiana, por lo que la prueba debe realizarse durante la mañana. Las muestras deben ser recolectadas en citrato y no en EDTA (ácido etilendiaminotetraacético) y debe evitarse el consumo de antiinflamatorios no esteroideos y antiagregantes por lo menos en los siete días previos a la realización de la prueba. No debe valorarse la agregabilidad plaquetaria dentro de los tres meses siguientes a un evento trombótico3.
La prueba de agregometría plaquetaria se diseñó inicialmente para valorar hipofunción plaquetaria en condiciones hemorrágicas. Detecta agregados de gran número de plaquetas, no pequeños; grupos puede ser subóptima para valorar hiperagregabilidad e hiperreactividad plaquetaria. La citometría de flujo podría aportar información acerca de los mecanismos (receptores de superficie) que median la hiperagregabilidad, mayor sensibilidad para detectar pequeñas agrupaciones de plaquetas. En la tabla 2 se encuentran los criterios para realizar el diagnóstico del síndrome de la plaqueta pegajosa.
Criterios diagnósticos del síndrome de la plaqueta pegajosa
| Diagnóstico sugestivo | - Historia de tromboembolia e hipercoagulabilidad plaquetaria en una sola concentración de un solo reactivo. |
| Diagnóstico confirmatorio | - Historia de tromboembolia. - Hipercoagulabilidad plaquetaria en dos o más concentraciones de un solo reactivo. - Historia de tromboembolia e hipercoagulabilidad plaquetaria en una o más concentraciones de ambos reactivos. |
Adaptada de: Medicina y Laboratorio 2014; 20(11-12)1,3.
Se ha demostrado que la antiagregación con ácido acetil salicílico en dosis bajas es costo-efectiva como medida de prevención primaria y secundaria en pacientes con síndrome de la plaqueta pegajosa para disminuir episodios trombóticos.
La dosis usual de ácido acetilsalicílico empleada es de 80 a 100mg/día. Su administración se asocia a normalización de la agregometría plaquetaria. En caso de recurrencia de eventos trombóticos la dosis puede titularse hasta 325mg/día1,22. La Aspirina ha demostrado ser segura e igualmente efectiva en mujeres en etapa preconcepcional y durante la gestación10.
Si hay contraindicación o fracaso terapéutico con ácido acetilsalicílico (recurrencia en menos del 5% de los casos), se ha descrito la antiagregación con clopidogrel 75mg/día como monoterapia o en antiagregación dual. La suspensión de los antiagregantes se relaciona con recaídas.
El síndrome de la plaqueta pegajosa no es una indicación de anticoagulación como tal y por lo tanto ésta solo debe prescribirse si existe otra patología concomitante que así lo amerite (síndrome antifosfolípido, resistencia a la proteína C activada, etc.)3.
ConclusionesEl síndrome de la plaqueta pegajosa es una trombofilia primaria o adquirida caracterizada por un trastorno cualitativo de las plaquetas, en el que hay hiperagregabilidad con epinefrina y ADP (adenosín difosfato); adicionalmente se ha descrito su clara asociación con manifestaciones clínicas como cefalea, abortos recurrentes, complicaciones obstétricas, así como manifestaciones trombóticas venosas y arteriales en pacientes jóvenes y en sitios inusuales.
Su prevalencia parece ser igual o incluso superior a la de otras condiciones de hipercoagulabilidad como el síndrome antifosfolípido.
En la literatura actual se describe una respuesta adecuada a la monoterapia con ácido acetilsalicílico (ASA) en la prevención primaria y secundaria de eventos trombóticos; sin embargo, también se han descrito casos de resistencia en los que ha sido necesario el uso de otros antiagregantes e incluso de anticoagulación, como lo fue en el caso que se describe en esta revisión. Casos como el que el describe en el texto, llevan a considerar la gran importancia de que el personal del área de la salud se familiarice con este trastorno para aumentar su reconocimiento y así implementar medidas de prevención primaria y secundaria en la población afectada, con lo que se impactará de forma positiva la morbimortalidad de esta patología.
FinanciaciónNinguna.
Conflicto de interesesNinguno.