




El síndrome de deleción 22q11.2 (22q11.2DS) se produce por microdeleciones del brazo largo del cromosoma 22 en la región q11.2. Después del síndrome de Down, es el segundo síndrome genético más común. En pacientes con esquizofrenia, el 22q11.2DS tiene una prevalencia del 2%, mientras que en personas con esquizofrenia seleccionadas por características físicas específicas, aumenta un 32-53%.
ObjetivoDescribir las generalidades del 22q11.2DS, sus características clínicas, los aspectos genético-moleculares y la frecuencia de la microdeleción de 22q11.2 en diferentes poblaciones.
MétodosSe hizo una revisión desde 1967 hasta 2013 en bases de datos de publicaciones científicas, orientada a recopilar artículos sobre el 22q11.2DS y su relación con la esquizofrenia.
ResultadosEl 22q11.2DS es una entidad genética que se asocia a un fenotipo variable relacionado con defectos congénitos en diferentes tejidos y órganos, así como a una alta frecuencia de trastornos psiquiátricos, particularmente la esquizofrenia. Se ha identificado alta prevalencia en grupos de personas con esquizofrenia seleccionadas por características sindrómicas comunes, como dificultades de aprendizaje, rasgos faciales típicos, anomalías palatales y defectos cardiacos congénitos. Las técnicas de FISH, qPCR, MLPA y, recientemente, aCGH y NGS se están usando para diagnosticar esta microdeleción.
ConclusionesEn la práctica clínica es importante tener presente que las personas con 22q11.2DS tienen alto riesgo de sufrir esquizofrenia, ya que la región 22q11.2 alberga genes candidatos relacionados con vulnerabilidad a esquizofrenia. Se considera que la concomitancia de esta enfermedad y 22q11.2DS representa un subtipo genético de esquizofrenia. Existen criterios fenotípicos claros y métodos citogenéticos y moleculares para diagnosticar a este grupo de pacientes y optimizar un abordaje multidisciplinario en su seguimiento.
The 22q11.2 deletion syndrome (22q11.2DS) is associated with the microdeletion of this chromosomal region, and represents the second most common genetic syndrome after Down's syndrome. In patients with schizophrenia, 22q11.2DS has a prevalence of 2%, and in selected groups can be increased to between 32-53%.
ObjectiveTo describe the generalities of 22q11.2DS syndrome as a genetic subtype of schizophrenia, its clinical characteristics, molecular genetic aspects, and frequency in different populations.
MethodsA review was performed from 1967 to 2013 in scientific databases, compiling articles about 22q11.2DS syndrome and its association with schizophrenia.
ResultsThe 22q11.2 DS syndrome has a variable phenotype associated with other genetic syndromes, birth defects in many tissues and organs, and a high rate of psychiatric disorders, particularly schizophrenia. Likewise, it has been identified in clinical populations with schizophrenia selected by the presence of common syndromic characteristics. FISH, qPCR and MLPA techniques, and recently, aCGH and NGS technologies, are being used to diagnose this microdeletion.
ConclusionsIt is important in clinical practice to remember that people suffering the 22q11.2DS have a high genetic risk for developing schizophrenia, and it is considered that the simultaneous presence of this disease and 22q11.2DS represents a genetic subtype of schizophrenia. There are clear phenotypic criteria, molecular and cytogenetic methods to diagnose this group of patients, and to optimize a multidisciplinary approach in their monitoring.
La prevalencia de esquizofrenia en la población general es de alrededor del 1% y presenta un tipo de herencia característica de enfermedades multifactoriales. En estudios de gemelos monocigóticos, el riesgo de sufrirla cuando uno de los gemelos la tiene es de un 50%; los familiares en primer grado de consanguinidad tienen un riesgo de un 5–16%; los de segundo grado, del 2–5% y de tercer grado, alrededor del 2%1–5. El síndrome de deleción 22q11.2 (22q11DS) es un síndrome genético común asociado con la microdeleción intersticial o pérdida submicroscópica de material genético en la región genómica 22q11.2, que puede estar asociado con un subtipo genético de esta enfermedad mental6.
El 22q11.2DS se caracteriza por un fenotipo variable que incluye problemas cognitivos y de comportamiento, anomalías congénitas cardiacas y fascies características; tiene una prevalencia del 2% de las personas con esquizofrenia6, y aumenta desde un 32%6 hasta un 53%5,6 si se selecciona una subpoblación de esquizofrénicos con características fenotípicas asociadas con el 22q11.2DS. Igualmente, se conoce también que un 10–30% de las personas con el 22q11.2DS sufren eventos psicóticos en la etapa adulta, frecuentemente esquizofrenia, por lo cual representa un importante factor de riesgo de sufrir esta enfermedad7.
En alrededor del 90% de los casos, el origen de la microdeleción 22q11.2 es una mutación de novo, lo que significa que ninguno de los padres es portador de la deleción8,9; en el 10% restante, la microdeleción se hereda de padres con la mutación pero con fenotipo menos intenso6. El 22q11.2DS tiene expresividad variable, que se solapa a otras entidades clínicas como el síndrome de Di George (DGS), el síndrome velocardiofacial (VCFS) y el síndrome de Takao, lo que hace que el diagnóstico clínico sea difícil y se produzca subdiagnóstico y, por lo tanto, se limite el abordaje de las posibles complicaciones clínicas cardiacas, renales, vertebrales, inmunitarias, endocrinas y mentales; esto también impide el asesoramiento genético que oriente a las familias sobre heredabilidad y riesgo de recurrencia.
Con el objetivo de describir el 22q11.2DS, su prevalencia, sus características clínicas, su asociación con esquizofrenia, los aspectos genéticos-moleculares, las técnicas de diagnóstico molecular y, finalmente, aspectos relevantes de asesoría genética, se realizó una revisión de artículos publicados desde 1967 hasta 2013 en bases de datos de publicaciones científicas.
MétodoSe realizó una búsqueda en bases de datos en línea de la iniciativa InterRed-Salud Programa de Acceso a la Investigación en Salud (HINARI), (http://www.who.int/hinari/en/), ScienceDirect (http://www.sciencedirect.com/), PubMed (http://www.ncbi.nlm.nih.gov/pubmed/), BioMed Central (http://www.biomedcentral.com/), Elsevier (http://www.elsevier.com/wps/find/journal_browse.cws_home) y Schizophrenia Research Forum (http://www.szgene.org/).
Se utilizaron los descriptores: 22q11.2 deletion syndrome; 22q11.2DS; Schizophrenia; Velo-cardio-facial syndrome (VCFS); DiGeorge syndrome (DGS); chromosome 22. Los artículos seleccionados se publicaron entre 1967 y 2013.
Los criterios de inclusión fueron: a) tipos de estudio: experimental, poblacional, de casos y controles y de prevalencia; b) pacientes con esquizofrenia y reporte de la microdeleción 22q11.2; c) pacientes con microdeleción 22q11.2, VCFS o DGS y que reporten esquizofrenia; d) con técnicas de diagnóstico citogenético y molecular; e) con información de asesoría genética a pacientes que tengan el 22q11.2DS, y f) publicaciones de cualesquiera año y población.
Síndrome de deleción del 22q11.2El 22q11.2DS se reportó por primera vez en la década de los años cincuenta6,10; fue completamente descrito hacia 19706,11 y se identificó más frecuentemente en la década de los noventa, gracias a la disponibilidad de estudios citogenéticos moleculares. Este síndrome es un trastorno multisistémico asociado con la microdeleción en el brazo largo de uno de los dos cromosomas 22 en la región q11.26,12,13. La hemicigosis, presencia de una sola copia de una región cromosómica, del 22q11.2 se ha asociado con diversos fenotipos, de los cuales Pinquier et al14 destacan el DGS, el (VCFS) y el síndrome Takao (anomalía conotroncal y de la cara); todos estos síndromes forman una unidad compleja con el 22q11.2DS, ya que comparten características clínicas y moleculares, con distintos grados de expresividad6,9,13,15–20. La prevalencia de la microdeleción 22q11.2 se ha estimado en la población mundial en 1/4.0009,16,21, lo cual hace que sea la microdeleción intersticial más frecuente y que el 22q11.2DS sea el segundo síndrome genético más prevalente, después del síndrome de Down6.
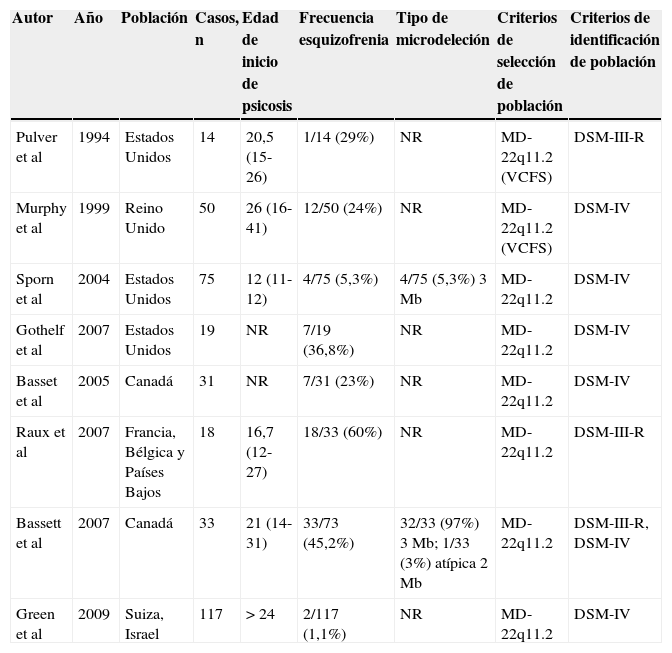
Los primeros estudios en reportar una posible asociación entre trastornos psiquiátricos y el 22q11.2DS se realizaron con personas con diagnóstico de VCFS. A partir de esta descripción, se ha reportado una alta prevalencia de síntomas psiquiátricos en pacientes con 22q11.2DS22,23, entre los que la esquizofrenia es el trastorno psiquiátrico más común22,24–26. Murphy et al27, en un estudio realizado a 40 adultos con 22q11.2DS, encontraron que el trastorno psiquiátrico más hallado, utilizando criterios DSM-IV, era la esquizofrenia, con una prevalencia del 25%, seguido por la depresión mayor (13%) y el trastorno bipolar (2,5%). Otro estudio identificó esquizofrenia en 4 de 14 adultos con 22q11.2DS (29%) utilizando criterios DSM-III-R23. Además, Pulver23 y Murphy28 plantean que el 85% de la población con VCFS tiene microdeleciones cromosómicas en 22q11.2 y presentan un 25% de probabilidad de padecer esquizofrenia (tabla 1). Según esos estudios, se estima que la probabilidad de que personas con 22q11.2DS sufran esquizofrenia es de 1 de cada 4 o 5 casos29, lo que indica que el riesgo de esquizofrenia en el 22q11.2DS es aproximadamente 25–30 veces mayor que el riesgo de la población general5,6,28,30–33 y el doble del riesgo para un familiar en primer grado de una persona con esquizofrenia6.
Frecuencia de casos con esquizofrenia entre los pacientes portadores de la microdeleción 22q11.2 o síndrome velocardiofacial (VCFS)
| Autor | Año | Población | Casos, n | Edad de inicio de psicosis | Frecuencia esquizofrenia | Tipo de microdeleción | Criterios de selección de población | Criterios de identificación de población |
|---|---|---|---|---|---|---|---|---|
| Pulver et al | 1994 | Estados Unidos | 14 | 20,5 (15-26) | 1/14 (29%) | NR | MD-22q11.2 (VCFS) | DSM-III-R |
| Murphy et al | 1999 | Reino Unido | 50 | 26 (16-41) | 12/50 (24%) | NR | MD-22q11.2 (VCFS) | DSM-IV |
| Sporn et al | 2004 | Estados Unidos | 75 | 12 (11-12) | 4/75 (5,3%) | 4/75 (5,3%) 3 Mb | MD-22q11.2 | DSM-IV |
| Gothelf et al | 2007 | Estados Unidos | 19 | NR | 7/19 (36,8%) | NR | MD-22q11.2 | DSM-IV |
| Basset et al | 2005 | Canadá | 31 | NR | 7/31 (23%) | NR | MD-22q11.2 | DSM-IV |
| Raux et al | 2007 | Francia, Bélgica y Países Bajos | 18 | 16,7 (12-27) | 18/33 (60%) | NR | MD-22q11.2 | DSM-III-R |
| Bassett et al | 2007 | Canadá | 33 | 21 (14-31) | 33/73 (45,2%) | 32/33 (97%) 3 Mb; 1/33 (3%) atípica 2 Mb | MD-22q11.2 | DSM-III-R, DSM-IV |
| Green et al | 2009 | Suiza, Israel | 117 | > 24 | 2/117 (1,1%) | NR | MD-22q11.2 | DSM-IV |
DSM: Manual diagnóstico y estadístico de los trastornos mentales; MD-22q11.2: microdeleción de 22q11.2; NR: no reporta; VCFS: síndrome velocardiofacial.
En pacientes con esquizofrenia, la microdeleción 22q11.2 puede tener una frecuencia aproximada del 2%6,25,31, y llega a ser 80 veces más alta que en la población general (0,025%); la prevalencia aumenta en población con esquizofrenia seleccionada con criterios fenotípicos asociados con el 22q11.2DS desde un 326 hasta un 53%5,6,30,34–36.
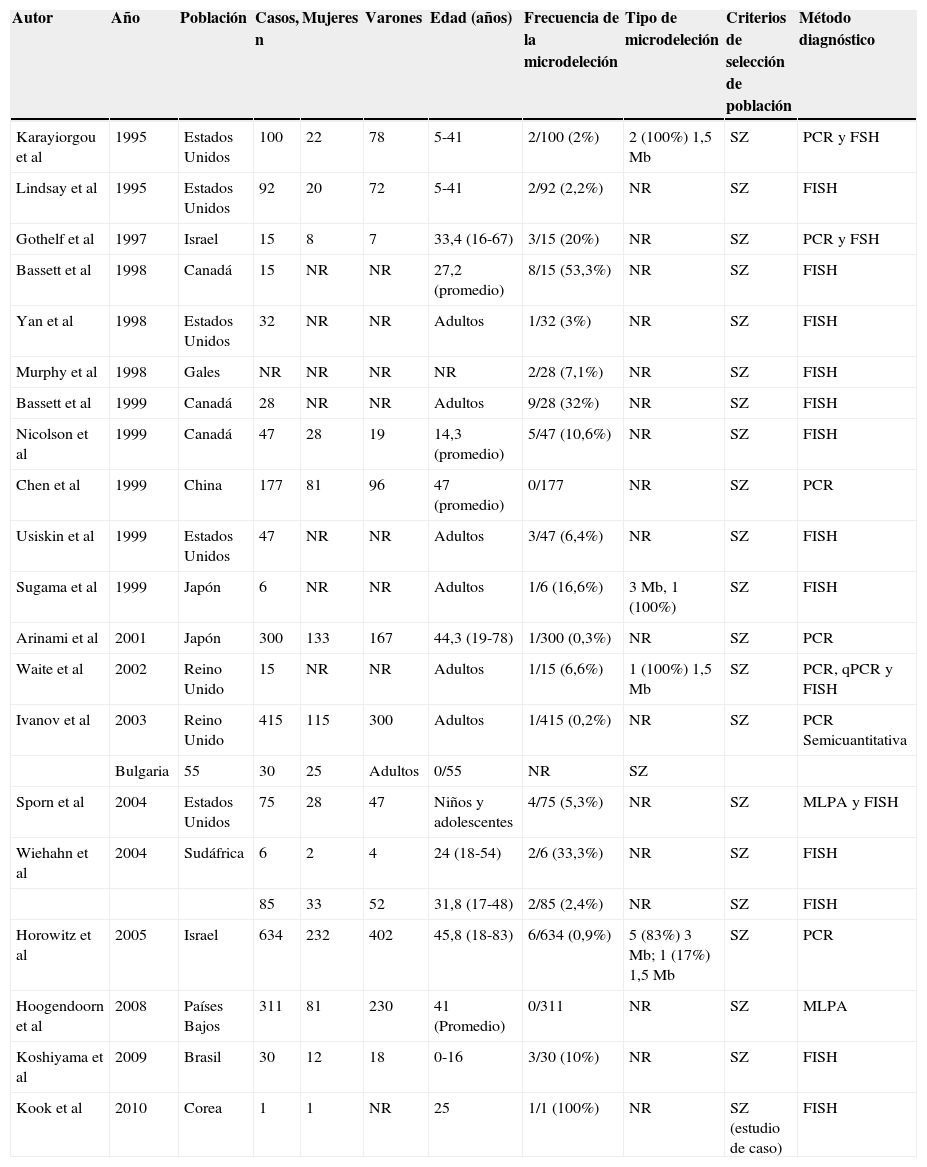
Karayiorgou et al25 encontraron que el 2% de una población adulta estadounidense con esquizofrenia, seleccionada al azar, presentó la microdeleción 22q11.2; posteriormente Yan et al37, que caracterizaron a sujetos con esquizofrenia desde la niñez, encontraron que el 3% de los pacientes presentaban esta microdeleción; asimismo, Wiehahn et al38 reportaron una prevalencia del 2,4% en una muestra de 85 pacientes con esquizofrenia; estudios complementarios en población joven con esquizofrenia de inicio en la infancia encontraron que la microdeleción podría ser incluso más frecuente, con prevalencias que oscilaron entre el 1,9 y el 11,939–42 (tabla 2). En contraste, no hay evidencia de mayor incidencia de esquizofrenia en otros síndromes genéticos con fenotipo conductual6,43. Es claro que las personas con el 22q11.2DS tienen alto riesgo de sufrir esta enfermedad9; por ello se considera que la concomitancia de esquizofrenia y 22q11.2DS representa un subtipo genético de esquizofrenia17.
Frecuencia de la microdeleción de 22q11.2 en pacientes con esquizofrenia
| Autor | Año | Población | Casos, n | Mujeres | Varones | Edad (años) | Frecuencia de la microdeleción | Tipo de microdeleción | Criterios de selección de población | Método diagnóstico |
|---|---|---|---|---|---|---|---|---|---|---|
| Karayiorgou et al | 1995 | Estados Unidos | 100 | 22 | 78 | 5-41 | 2/100 (2%) | 2 (100%) 1,5 Mb | SZ | PCR y FSH |
| Lindsay et al | 1995 | Estados Unidos | 92 | 20 | 72 | 5-41 | 2/92 (2,2%) | NR | SZ | FISH |
| Gothelf et al | 1997 | Israel | 15 | 8 | 7 | 33,4 (16-67) | 3/15 (20%) | NR | SZ | PCR y FSH |
| Bassett et al | 1998 | Canadá | 15 | NR | NR | 27,2 (promedio) | 8/15 (53,3%) | NR | SZ | FISH |
| Yan et al | 1998 | Estados Unidos | 32 | NR | NR | Adultos | 1/32 (3%) | NR | SZ | FISH |
| Murphy et al | 1998 | Gales | NR | NR | NR | NR | 2/28 (7,1%) | NR | SZ | FISH |
| Bassett et al | 1999 | Canadá | 28 | NR | NR | Adultos | 9/28 (32%) | NR | SZ | FISH |
| Nicolson et al | 1999 | Canadá | 47 | 28 | 19 | 14,3 (promedio) | 5/47 (10,6%) | NR | SZ | FISH |
| Chen et al | 1999 | China | 177 | 81 | 96 | 47 (promedio) | 0/177 | NR | SZ | PCR |
| Usiskin et al | 1999 | Estados Unidos | 47 | NR | NR | Adultos | 3/47 (6,4%) | NR | SZ | FISH |
| Sugama et al | 1999 | Japón | 6 | NR | NR | Adultos | 1/6 (16,6%) | 3 Mb, 1 (100%) | SZ | FISH |
| Arinami et al | 2001 | Japón | 300 | 133 | 167 | 44,3 (19-78) | 1/300 (0,3%) | NR | SZ | PCR |
| Waite et al | 2002 | Reino Unido | 15 | NR | NR | Adultos | 1/15 (6,6%) | 1 (100%) 1,5 Mb | SZ | PCR, qPCR y FISH |
| Ivanov et al | 2003 | Reino Unido | 415 | 115 | 300 | Adultos | 1/415 (0,2%) | NR | SZ | PCR Semicuantitativa |
| Bulgaria | 55 | 30 | 25 | Adultos | 0/55 | NR | SZ | |||
| Sporn et al | 2004 | Estados Unidos | 75 | 28 | 47 | Niños y adolescentes | 4/75 (5,3%) | NR | SZ | MLPA y FISH |
| Wiehahn et al | 2004 | Sudáfrica | 6 | 2 | 4 | 24 (18-54) | 2/6 (33,3%) | NR | SZ | FISH |
| 85 | 33 | 52 | 31,8 (17-48) | 2/85 (2,4%) | NR | SZ | FISH | |||
| Horowitz et al | 2005 | Israel | 634 | 232 | 402 | 45,8 (18-83) | 6/634 (0,9%) | 5 (83%) 3 Mb; 1 (17%) 1,5 Mb | SZ | PCR |
| Hoogendoorn et al | 2008 | Países Bajos | 311 | 81 | 230 | 41 (Promedio) | 0/311 | NR | SZ | MLPA |
| Koshiyama et al | 2009 | Brasil | 30 | 12 | 18 | 0-16 | 3/30 (10%) | NR | SZ | FISH |
| Kook et al | 2010 | Corea | 1 | 1 | NR | 25 | 1/1 (100%) | NR | SZ (estudio de caso) | FISH |
FISH: hibridación in situ con fluorescencia; MLPA: amplificación de sondas dependiente de ligandos múltiples; NR: no reporta; PCR: reacción en cadena de la polimerasa; SZ: esquizofrenia.
El 22q11.2DS presenta un fenotipo variable que está relacionado con la edad de los individuos44 y con la presencia de otros síndromes genéticos, como el VCFS y el DGS44–47.
Se han identificado más de 180 características clínicas en este síndrome48,49 y, debido a la variabilidad significativa en la expresión del fenotipo, en muchos casos el diagnóstico puede pasarse por alto, con un mayor subdiagnóstico en adultos con aparición tardía de síntomas psiquiátricos6,9,15,21,50,51.
La expresión clínica del 22q11.2DS está asociada a menudo con una alta frecuencia de defectos congénitos que afectan a un gran número de órganos32,52; las características clínicas más comunes son dismorfismo facial50, paladar hendido, defectos congénitos del corazón, discapacidades del aprendizaje y alta frecuencia de trastornos psiquiátricos, particularmente esquizofrenia6,17,27,53; la frecuencia de estas características es incierta, ya que las prevalencias varían según la edad51,54–56 y además varían entre familias e incluso entre los miembros de la misma familia57,58; algunas características, como defectos cardiacos y la hipocalcemia, se pueden diagnosticar en la infancia, otros por lo general se identifican más tarde, como dificultades de aprendizaje, paladar hendido y habla hipernasal, a menudo asociada con insuficiencia velofaríngea11.
El 22q11.2DS se puede identificar en poblaciones clínicas con esquizofrenia seleccionadas por la presencia de características sindrómicas comunes26,50. Bassett et al6 proponen ciertos criterios de criba enmarcados en el 22q11.2DS que incrementan la probabilidad de identificar la microdeleción en poblaciones con esquizofrenia; ésta puede sospecharse en casos que presenten por lo menos dos de las siguientes características:
- •
Problemas de comportamiento.
- •
Habla hipernasal, historia de terapia del lenguaje, incompetencia velofaríngea o paladar hendido (usualmente submucoso).
- •
Rasgos faciales: cara estrecha y alargada, fisuras palpebrales estrechas, pómulos hipoplásicos, nariz prominente, orejas pequeñas, boca pequeña y/o retrognatia.
- •
Dificultades del aprendizaje, historia de necesidad de educación especial, retardo mental.
- •
Defectos congénitos del corazón: defecto septal ventricular, tetralogía de Fallot, arco aórtico derecho, doble arco aórtico.
- •
Otras anormalidades congénitas: polidactilia, escoliosis, anomalía renal, hipospadias.
- •
Historia de hipocalcemia (neonatal, niñez, adolescencia, o adultez temprana) y/o hipoparatiroidismo.
- •
Atimia o inmunodeficiencia grave en la infancia.
Las frecuencias más altas de la microdeleción 22q11.2 oscilan entre el 14 y el 53%7 y se han reportado en los estudios que seleccionaron a pacientes con esquizofrenia que tienen varias de las características clínicas del VCFS26,39,59. Entre esos estudios se cita a Bassett et al26, que reportaron una asociación del VCFS con trastornos psicóticos en adultos caucásicos en Canadá; en ese estudio se evaluó a 15 sujetos diagnosticados de esquizofrenia mediante criterios DSM-IV o con trastorno esquizoafectivo referidos con dos o más características asociadas con el 22q11.2DS (cardiacas, faciales u otras anomalías congénitas y/o dificultades de aprendizaje); esos pacientes no tenían ningún tipo de diagnóstico previo asociado a VCFS; 8 fueron diagnosticados de VCFS en el transcurso de la evaluación, por lo que se destaca que la tasa de asociación de la deleción 22q11.2 encontrada en el estudio en pacientes con esquizofrenia y rasgos característicos del 22q11.2DS fue de 8/15 (53,3%).
Este hallazgo es comparable con lo reportado en otros estudios de pacientes con características sindrómicas. Gothelf et al59 diagnosticaron la microdeleción 22q11.2 en 15 pacientes israelíes con esquizofrenia y características sindrómicas relacionadas con esta alteración, todos sin sospecha previa del VCFS. La microdeleción estuvo presente en 3/15 pacientes (20%). De manera similar, Bassett et al6 reportan la presencia de la microdeleción 22q11.2 en 9/28 pacientes (32%) con esquizofrenia referidos por fuentes psiquiátricas por presentar dos o más de las características relacionadas con 22q11.2DS (tabla 2).
Aspectos genéticos-moleculares de la microdeleción 22q11.2En la mayoría de los casos (90%), la deleción se presenta por una mutación de novo12,32,60, que puede producirse durante la gametogénesis. En el 10% restante, se hereda por transmisión autosómica dominante de un padre afectado que puede presentar solo manifestaciones leves60,61. Este patrón de herencia lo sospecharon inicialmente Shprintzen et al. en 1978, después de observar los casos de VCFS con la transmisión de madre a hija, y se confirmó en 1985, con la descripción de los primeros casos de transmisión de padres a hijos62,63.
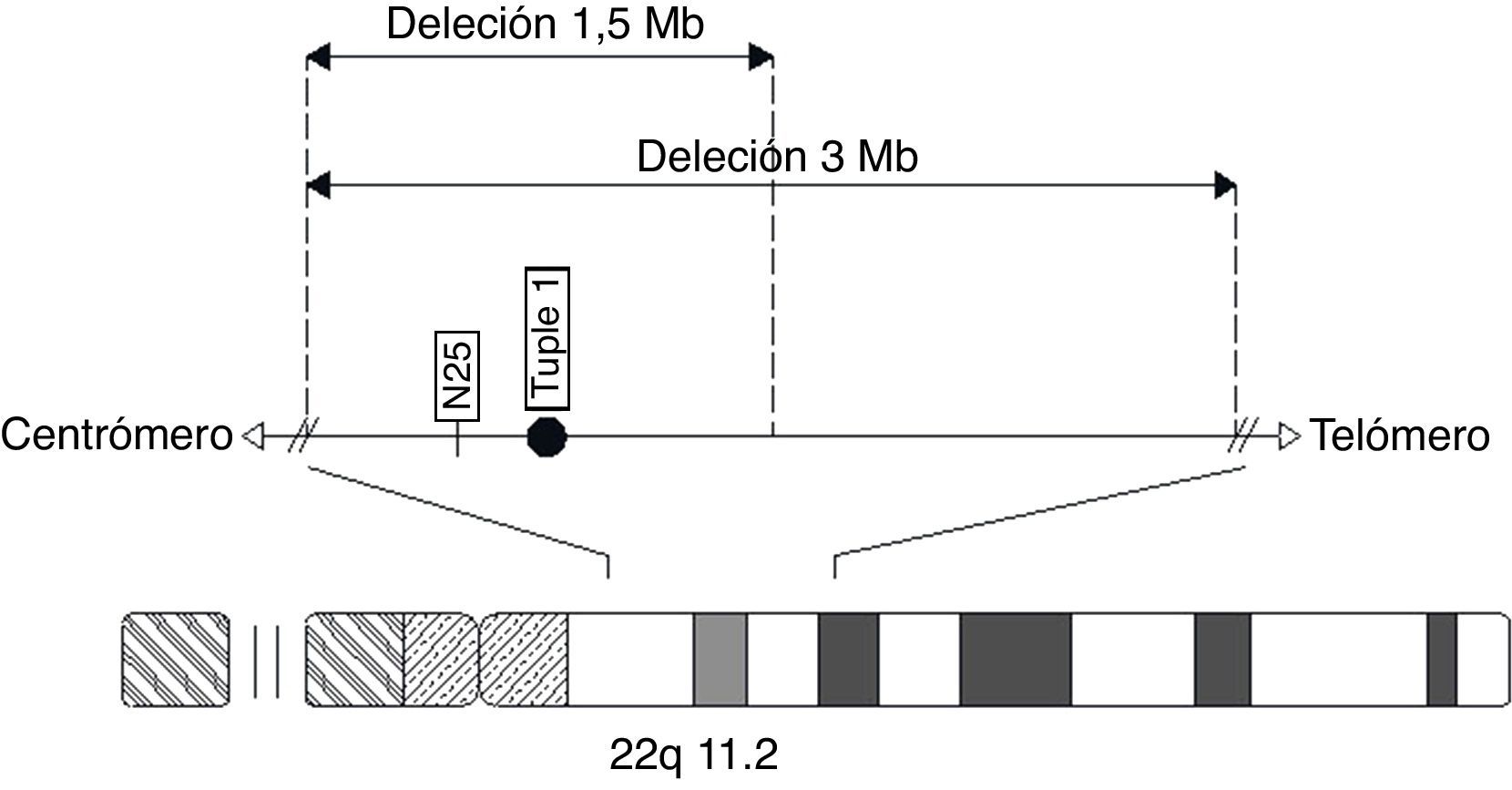
La región 22q11.2 contiene regiones de repeticiones de bajo número de copias (LCR22) que probablemente predisponen a alteraciones en la recombinación meiótica y a ulteriores reordenamientos estructurales en esta región64,65. Existe tres tipos de microdeleción 22q11.2: el tipo I, la más común, corresponde a la mayor deleción, estimada de 3 Mb66; el tipo II es una deleción más pequeña que se solapa a la región proximal de la región de deleción tipo I con un tamaño aproximado de 1,5 Mb (ambos tipos se han reportado en esquizofrenia25,32,51,53,64,67), y el tipo III, la deleción más pequeña, que se solapa al extremo distal de la deleción más larga (tipo I) y se suele considerar una deleción atípica que no se relaciona con esquizofrenia67,68. También se han encontrado otras deleciones de diversas extensiones. Es interesante que no haya una región cromosómica en hemicigosis común en todas las personas con 22q11.2DS y esquizofrenia9,66 (figura 1), lo que indica que en la región 22q11.2 hay más de un gen involucrado en el incremento de la susceptibilidad a la enfermedad mental28. El descubrimiento de estos genes podría tener un papel importante en mejorar la comprensión de la etiología molecular de esta enfermedad51.
 y la tipo II de 1,5 Mb, ambas relacionadas con esquizofrenia. También la distribución de las sondas comúnmente usadas para la detección de la región q11.2 (TUPLE 1 y N25) (basado en Karayiorgou et al25, Ivanov et al40, Kurahashi et al67, Weksberg et al84, Waite et al85, Carlson et al94, Pereira et al95, Gioli–Pereira et al96, Nogueira et al97 y Garavelli et al98).")
Esquema del cromosoma 22, que muestra la localización de los tipos de microdeleción tipo I de 3 Mb (la más común) y la tipo II de 1,5 Mb, ambas relacionadas con esquizofrenia. También la distribución de las sondas comúnmente usadas para la detección de la región q11.2 (TUPLE 1 y N25) (basado en Karayiorgou et al25, Ivanov et al40, Kurahashi et al67, Weksberg et al84, Waite et al85, Carlson et al94, Pereira et al95, Gioli–Pereira et al96, Nogueira et al97 y Garavelli et al98).
Investigaciones relacionadas con posibles factores de riesgo genético de sufrir esquizofrenia —como mutaciones y polimorfismos de un solo nucleótido (SNP)— se han registrado en bases de datos genómicas. Una de las bases de datos más reconocidas es la SzGene database (http://www.szgene.org/), una confiable herramienta en línea que, mediante estudios de metanálisis, logra hacer una integración estructurada y sistemática de la evidencia acumulada de estudios genéticos de asociación en esquizofrenia69,70.
La SzGene database muestra que, en la región 22q11.2, se han descrito genes candidatos para la esquizofrenia; algunos codifican receptores de neurotransmisores o enzimas involucradas en su metabolismo69,70. Investigaciones en 22q11.2DS se han centrado principalmente en la participación de los genes catecol-O-metil transferasa (COMT) y prolina deshidrogenasa (PRODH) en la neurobiología del 22q11.2DS66,71,72. La enzima codificada por el gen COMT tiene un papel crucial en el metabolismo del neurotransmisor dopamina, especialmente en la corteza prefrontal6,31. Se cree que la función anormal de las vías dopaminérgicas tienen un papel importante en la esquizofrenia73. La deleción homocigótica del gen PRODH que codifica la prolina deshidrogenasa, enzima mitocondrial que cataliza la conversión de la prolina a glutamato, conduce a cantidades de prolina significativamente elevadas71,74; el aumento de la concentración plasmática de prolina se ha observado en poblaciones de pacientes psicóticos75–78 y se ha asociado con problemas neurológicos en el 22q11.2DS74,75,79,80. También se ha planteado la hipótesis de interacción epistática entre estos dos genes, ya que ambos convergen funcionalmente en el sistema dopaminérgico81. Así, en el contexto de la microdeleción 22q11.2, la reducción simultánea de la actividad tanto de COMT y PRODH puede conducir a una desregulación sinérgica de los sistemas dopaminérgicos que resulta en un estado hiperdopaminérgico, que puede predisponer a psicosis y esquizofrenia75,81.
Todo lo anterior respalda que la región 22q11.2 alberga genes implicados en la esquizofrenia, y es posible que el mayor riesgo asociado con esta microdeleción se deba a la contribución de más de un gen ligado físicamente al lugar de la microdeleción25,51. La tabla 3 muestra los genes candidatos destacados en la esquizofrenia que refiere la SzGene database en la región 22q11.2.
Genes candidatos destacados en la esquizofrenia ubicados en la región 22q11.2
| Gen (sigla en inglés) | Posición genómica (GRCh38/hg38) | Proteína codificada |
|---|---|---|
| DGCR2 | chr22:19,036,282-19,122,454 | SDG región crítica proteína 2 |
| DGCR5 | chr22:18,970,498-18,994,629 | SDG región crítica 5 (no codifica para proteína) |
| DGCR6 | chr22:20,314,238-20,320,105 | SDG región crítica proteína 6 |
| GNB1L | chr22:19,788,411-19,854,939 | Proteína similar a la subunidad beta de la proteína de unión a guanina a nucleótidos de guanina (proteína G) |
| PRODH | chr22:18,912,774-18,936,553 | Prolina deshidrogenasa (oxidasa) 1 |
| TUBA8 | chr22:18,110,687-18,131,731 | Tubulina, alfa 8 |
| UFD1L | chr22:19,449,941-19,479,215 | Proteína similar la de fusión y degradación de ubiquitina 1 |
| ARVCF | chr22:19,969,879-20,016,786 | Proteína de repetición de armadillo |
| CLDN5 | chr22:19,523,024-19,525,337 | Claudin 5 |
| COMT | chr22:19,962,547-19,969,975 | Catecol-O-metiltransferasa |
| HTF9C | chr22:20112566-20116906 | HpaII diminutos fragmentos locus 9C |
| PCQAP | chr22:20507679-20587617 | Proteína asociada a cofactor positivo 2, glutamina/Q–rica |
| RANBP1 | chr22:20,117,953-20,127,357 | Proteína de unión RAN 1 |
| RTN4R | chr22:20,241,415-20,268,293 | Precursor del receptor retículon 4 |
| TBX1 | chr22:19,756,703-19,783,589 | T-box 1 |
| ZDHHC8 | chr22:23,390,605-23,402,612 | Dedo de cinc, contiene ocho dominios DHHC |
| PIK4CA | chr22:20707702-20858782 | Fosfatidilinositol cinasa 4, catalítico |
| SNAP29 | chr22:20,859,004-20,891,213 | Proteína asociada a sinaptosomas, 29 kDa |
| GSTT1 | chr22:23,957,816-23,961,149 | Glutatión S-transferasa theta 1 |
SDG: síndrome de DiGeorge.
La microdeleción 22q11.2 se puede identificar mediante técnicas citogenéticas moleculares como la hibridación in situ con fluorescencia (FISH)25,30,34,38,40,41,53,82,83, y técnicas moleculares como la reacción en cadena de la polimerasa (PCR)40,84 y algunas de sus variantes, como PCR cuantitativa (qPCR) y PCR multiplex dependiente de ligación (MLPA)20,41,42,53,84.
Para FISH se usa una sonda que es una secuencia de cadena sencilla de ADN marcada con fluorescencia. La secuencia de la sonda es complementaria a la secuencia del ADN que se quiere identificar. En la forma más sencilla de esta técnica, se permite que las células proliferen y se las detiene en la metafase de la mitosis, que es el momento en que los cromosomas están más condensados. Después, las metafases se someten a hibridación con la sonda, y por complementariedad de pares de bases, esta se unirá a la región genómica de interés. En un genoma normal se identifican dos señales: una por cada región de cada cromosoma homologo. Si hay una deleción, la sonda no puede unirse y, por lo tanto, la señal fluorescente correspondiente no se produce. Para detectar la microdeleción 22q11.2, se usan la sondas N25 o TUPLE1, que reconocen secuencias de ADN ubicadas en las regiones más frecuentemente perdidas en este síndrome25,85.
También se puede diagnosticar la microdeleción usando la PCR. Con esta es posible amplificar millones de veces una región genómica específica. También se puede cuantificar el producto amplificado, que depende del número de copias de ADN con que se inicia la reacción. Así, si se trata de una muestra de ADN procedente de una persona sin deleción, se partirá de dos copias de la misma región por cada célula. En el caso de una persona con deleción, se partirá de una sola copia de ADN por célula, por que si se compara la cantidad de producto amplificado entre una persona normal con una con el síndrome de deleción, esta tendrá la mitad que la persona sin deleción25,40,85.
Aunque FISH, qPCR y MLPA son los procedimientos estándar y más accesibles para el diagnóstico rápido de la microdeleción 22q11.2, es importante mencionar que recientemente se han desarrollado nuevas técnicas caracterizadas por su alta resolución y sensibilidad que también permiten identificar la microdeleción. Estas son la hibridación genómica comparativa en formato de microarreglos (conocida por las siglas aCGH) y la secuenciación masiva de nueva generación (NGS)86–89.
Síndrome de deleción de 22q11.2 y asesoramiento genéticoEl diagnóstico de la microdeleción 22q11.2 cambia significativamente el consejo genético y el tratamiento de los pacientes. El diagnóstico temprano permite influir en la evolución de la enfermedad y optimizar los resultados para un manejo adecuado de la condiciones asociadas con ella53,90. Los portadores de la microdeleción esencialmente tienen que ser evaluados multidisciplinariamente de manera regular, teniendo en cuenta no solo el manejo médico, sino el psiquiátrico, genético, psicológico y lingüístico12,53. Diferentes estudios reportan que en adultos el tratamiento se ve afectado por el aislamiento social, tendencias a la pasividad, y en el caso de trastornos psiquiátricos, en ocasiones solo hay respuesta parcial a los medicamentos antipsicóticos12.
El manejo de los trastornos psiquiátricos de los portadores de la microdeleción 22q11.2 presenta dificultades debido a los estigmas asociados con las enfermedades mentales, como lo manifiesta un estudio realizado por Martin et al29, en el que se encuestó a médicos genetistas en Canadá acerca del abordaje y las perspectivas de manejo en materia de divulgación de las manifestaciones clínicas del 22q11.2DS, particularmente en cuanto al riesgo de enfermedad psiquiátrica.
Dicho estudio mostró que cuando la microdeleción 22q11.2 se diagnostica en la infancia, es común que no se informe inmediatamente a los padres del riesgo aumentado de enfermedades psiquiátricas, sino que se hace en diferentes momentos de la vida del paciente. Aunque los médicos genetistas coinciden en que es importante revelar el aumento del riesgo de sufrir una enfermedad psiquiátrica, los encuestados informan que la discusión de temas psiquiátricos con los padres es difícil; por el contrario, otras anormalidades médicas asociadas a la microdeleción se informan desde el diagnóstico en la infancia.
El consejo genético oportuno permite brindar a los padres el conocimiento sobre riesgos de recurrencia de la enfermedad en futuras generaciones, así como enfatizar a los padres la importancia de llevar a cabo un manejo adecuado de los portadores de la microdeleción 22q11.2, quienes enfrentan un riesgo alto de psicosis en la edad adulta91.
DiscusiónEn la práctica clínico-psiquiátrica, dos aspectos respaldan la necesidad de seguimiento monitoreo y diagnóstico de posibles casos portadores de la microdeleción 22q11.2: el primero es que las personas con esta microdeleción tienen un factor de riesgo genético importante, que puede estar asociado con el desencadenamiento de la esquizofrenia, y en segundo lugar, que dentro de la población de personas con esquizofrenia, existe un grupo de pacientes que sufren el 22q11.2DS y que en algunos casos puede estar subdiagnosticado.
En relación con el primer aspecto, se debe tener presente que al estar establecido que el 22q11.2DS es un factor de riesgo genético de esquizofrenia, la cual se desarrolla en un 10–30% de los portadores de la microdeleción 22q11.26,7,31, es importante realizar un seguimiento neuropsiquiátrico de estas personas que considere los diferentes factores predisponentes y precipitantes asociados con el desarrollo de este trastorno mental.
La identificación de los síntomas precursores de un trastorno psicótico futuro para las personas en riesgo proporciona una oportunidad para la intervención temprana en las personas con síntomas psicóticos92 y, debido al alto riesgo de psicosis en la edad adulta91, el tratamiento de la psicopatía infantil podría ser crucial en la mitigación de los riesgos de las enfermedades psiquiátricas.
Schneider et al12 afirman que el apoyo regular para el paciente y su familia por el médico general y un psiquiatra con al menos una evaluación anual del funcionamiento psicológico evita cuestiones emergentes, y una colaboración estrecha con los padres maximiza las posibilidades de una autonomía adulta. Asimismo, recibir asesoramiento genético adecuado, que aborde las implicaciones clínicas y los riesgos de heredabilidad debido a la demostrada transmisión autosómica dominante de esta alteración, contribuiría a liberar a los padres de la culpa inapropiada por las manifestaciones conductuales de la enfermedad43.
Es importante profundizar en los factores de riesgo genéticos implicados en la esquizofrenia32 y en la heterogeneidad fenotípica asociada con el 22q11.2DS, que permita realizar su diagnóstico temprano en la población de riesgo6. Los estudios biológicos y moleculares actuales sobre los genes de susceptibilidad ubicados en la región delecionada posiblemente aporten a la identificación de predictores tempranos de trastornos psiquiátricos de inicio tardío32, así como la evaluación del curso del fenotipo psiquiátrico92.
En referencia al segundo aspecto, como el 22q11.2DS es un subtipo genético de esquizofrenia, es importante tener en cuenta en la evaluación psiquiátrica que, para los pacientes con esquizofrenia que cumplan al menos dos de los criterios de criba enmarcados en el 22q11.2DS6, es recomendable un estudio genético que confirme la sospecha de una posible microdeleción de 22q11.2. En Colombia está disponible este tipo de estudios tanto citogenéticos como moleculares.
En los reportes de Bassett et al26,51 y Gothelf et al93, se muestran fotografías de pacientes esquizofrénicos portadores de la microdeleción de 22q11.2; es evidente la amplia variabilidad en la expresión del síndrome y en la manifestación de las características físicas asociadas32.
Aunque la literatura mundial ha establecido que el 22q11.2DS tiene una prevalencia del 2% de los pacientes con esquizofrenia, entre las personas con esquizofrenia seleccionadas por características físicas específicas aumenta un 32–53%, sería importante realizar a escala nacional estudios transversales y longitudinales que permitan conocer la frecuencia de este síndrome en la población colombiana para comprender mejor los trastornos psiquiátricos asociados con el 22q11.2DS y contribuir al manejo multidisciplinario de los individuos afectados.
Un alto índice de sospecha y una evaluación periódica multidisciplinaria pueden ayudar a identificar a los pacientes con esquizofrenia y deleción de 22q11.2, para una posterior intervención de los individuos afectados.
ConclusionesCon esta revisión, se resalta la importancia de tener presente en la práctica clínica que las personas con el 22q11.2DS tienen alto riesgo genético de sufrir esquizofrenia; además, se considera que la concomitancia de esta enfermedad y el 22q11.2DS representa un subtipo genético de esquizofrenia. Igual que existen criterios fenotípicos claros y métodos diagnósticos citogenéticos y moleculares, muchos de ellos disponibles en nuestro país para diagnosticar a este grupo de pacientes y optimizar un abordaje multidisciplinario que permita mejorar su seguimiento y orientar a la familia sobre las implicaciones clínicas y riesgos de heredabilidad de esta enfermedad. Lo más conveniente para los pacientes es lograr un diagnóstico temprano, pues influye en la evolución de la enfermedad y optimiza los resultados en cuanto al manejo adecuado de las condiciones asociadas con este subtipo genético de esquizofrenia.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
