



La miositis por cuerpos de inclusión forma parte del grupo de las miopatías inflamatorias, de las que representa el 30%; es considerada una enfermedad huérfana, ya que se estima que su prevalencia es menor a 5 por cada 10.000 habitantes. Produce debilidad y atrofia de los músculos proximales y distales. Los mecanismos fisiopatológicos son principalmente autoinmunes, inflamatorios y degenerativos. Se presentan 2 casos de mujeres, quienes acudieron a urgencias por pérdida progresiva de la fuerza en miembros superiores e inferiores, con debilidad muscular asimétrica de curso progresivo.
Inclusion body myositis is part of the group of inflammatory myopathies, representing 30% of this group of diseases, and is considered an orphan disease because its estimated prevalence is less than 5 per 10,000 inhabitants. It produces weakness and atrophy of the proximal and distal muscles. The pathophysiological mechanisms are mainly autoimmune, inflammatory, and degenerative. The cases are presented on 2 female patients who came to the emergency department due to progressive loss of upper and lower limb strength, and progressive asymmetric muscle weakness.
El concepto miopatía de cuerpos de inclusión (MCI) fue descrito por primera vez en 1971 y adoptado después de la descripción de casos de pacientes con polimiositis resistente a tratamiento convencional (corticosteroides). En 1995 se dieron a conocer los primeros criterios diagnósticos de esta enfermedad1.
La MCI es un trastorno esporádico poco frecuente, incluido dentro del grupo de miopatías inflamatorias idiopáticas. Se estima que su prevalencia es de 5-9 casos por millón de adultos, y que es la miopatía adquirida más frecuente en mayores de 50 años (edad de inicio promedio, 60 años) con afección mayor en mujeres2.
Respecto a la fisiopatología, se cree que el envejecimiento asociado a la acumulación y sedimentación de proteínas en el entorno intracelular desempeña un papel en la patogénesis, si bien parece que el estrés oxidativo y la reacción al estrés en el retículo endoplásmico también contribuyen. Los factores anteriores conducen de forma acumulativa a la degeneración progresiva y a la necrosis del músculo. Se han descrito 3 mecanismos principales de lesión: autoinmune, inflamatorio y degenerativo. Las células T citotóxicas desempeñan un papel importante, puesto que invaden y destruyen las fibras musculares, principalmente aquellas que son vacuoladas o con depósitos amiloides y generan, adicionalmente, daños neurodegenerativos2-5.
La principal manifestación clínica es la debilidad generalizada de curso insidioso, de compromiso tanto distal como proximal, con predominio del último, expresado por el paciente como incapacidad para levantarse de la silla o como caídas frecuentes. Al examen físico la principal característica es la debilidad de los músculos flexores de los dedos a nivel distal6,7.
Hallazgos clínicos que se diferencian de otras enfermedades del mismo grupo (dermatomiositis o polimiositis) son el compromiso distal y asimétrico, la presencia de mialgias de intensidad leve, mayor atrofia esperada de acuerdo con el tiempo del cuadro clínico y, raramente, disfagia severa8.
El hallazgo de enzimas musculares elevadas es la principal anormalidad de laboratorio en estos pacientes. La elevación leve de la creatinina cinasa menor a 10 veces el valor normal es el principal diferenciador de otras miopatías inflamatorias. Hallazgos en electromiografía corresponden a miopatía inflamatoria. Los anticuerpos específicos para miositis suelen estar ausentes en la MCI. Las características patológicas típicas de esta entidad son: vacuolas anilladas con fibras musculares atróficas o normales, infiltrados inflamatorios y linfocitos CD8+ T9.
Los primeros criterios clínicos aceptados fueron los propuestos por Griggs en 1995; posteriormente, en el 2011, el European Neuromuscular Centre definió unos criterios para clasificar la enfermedad en 2 categorías: MCI clínico-patológica y MCI clínica1.
El tratamiento óptimo de la MCI actualmente es desconocido y la mayoría de las intervenciones han tenido beneficios limitados, por lo cual se obliga a la utilización de terapia inmunosupresora, con un pronóstico desfavorable si se compara con otras miopatías inflamatorias10.
Se presentan 2 casos de mujeres mayores de 50 años, quienes acudieron a urgencias por pérdida de la fuerza muscular en miembros superiores e inferiores, de tipo asimétrico, de curso progresivo, en las que se logró un diagnóstico histopatológico y clínico de MCI. Dada que esta entidad es poco frecuente en nuestro medio, se decidió reportar ambos casos en la literatura.
Caso clínico 1Mujer de 55 años, ama de casa, diestra, con antecedente de hipertensión arterial sistémica e hipotiroidismo primario controlado, quien acudió a urgencias por cuadro clínico de 4 meses de evolución consistente en pérdida progresiva de la fuerza muscular en cintura pélvica y escapular. La debilidad progresó de forma insidiosa en los últimos 20 días hasta llevarla a requerir asistencia para sus actividades básicas de la vida diaria. En la revisión por sistemas se documentó, en el último mes, una pérdida de alrededor de 10kg de peso, no intencional, asociada a hiporexia progresiva. Negó diaforesis, picos febriles, lesiones cutáneas u otros síntomas concomitantes. No había antecedentes familiares positivos para enfermedades autoinmunes. Al examen físico de ingreso tenía incapacidad para levantar los brazos por encima de los hombros y para levantarse de una silla, la fuerza muscular era 3/5 en todas las extremidades, excepto en miembro inferior izquierdo, en el que tenía 2/5. Los reflejos osteotendinosos eran normales, no había alteraciones sensitivas.
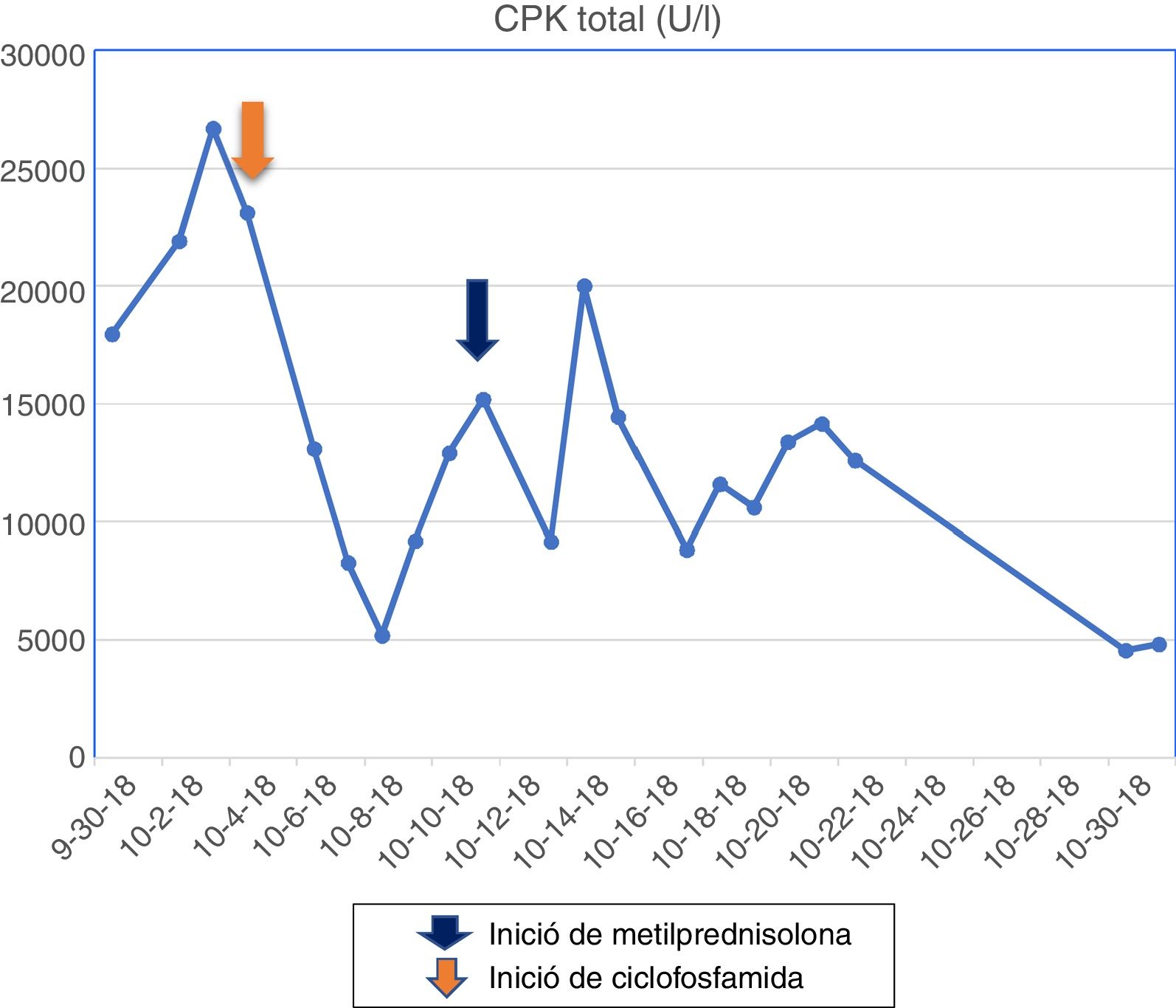
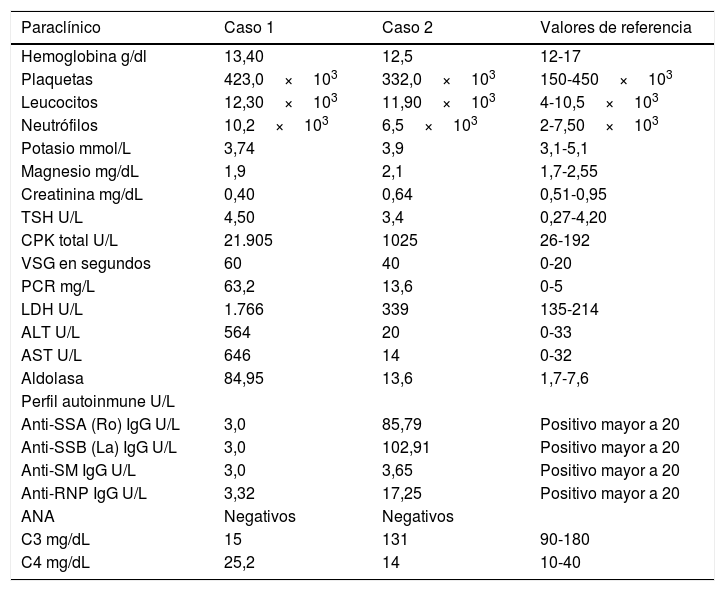
La paciente tenía formulación crónica de 20mg de lovastatina por 2 años como prevención primaria, sin conocer las razones para su prescripción, que en el contexto de la miopatía fue suspendida. Los paraclínicos de ingreso se observan en la tabla 1 y los cambios en la curva de la CPK total se aprecian en la figura 1.
Características serológicas de los pacientes tratados
| Paraclínico | Caso 1 | Caso 2 | Valores de referencia |
|---|---|---|---|
| Hemoglobina g/dl | 13,40 | 12,5 | 12-17 |
| Plaquetas | 423,0×103 | 332,0×103 | 150-450×103 |
| Leucocitos | 12,30×103 | 11,90×103 | 4-10,5×103 |
| Neutrófilos | 10,2×103 | 6,5×103 | 2-7,50×103 |
| Potasio mmol/L | 3,74 | 3,9 | 3,1-5,1 |
| Magnesio mg/dL | 1,9 | 2,1 | 1,7-2,55 |
| Creatinina mg/dL | 0,40 | 0,64 | 0,51-0,95 |
| TSH U/L | 4,50 | 3,4 | 0,27-4,20 |
| CPK total U/L | 21.905 | 1025 | 26-192 |
| VSG en segundos | 60 | 40 | 0-20 |
| PCR mg/L | 63,2 | 13,6 | 0-5 |
| LDH U/L | 1.766 | 339 | 135-214 |
| ALT U/L | 564 | 20 | 0-33 |
| AST U/L | 646 | 14 | 0-32 |
| Aldolasa | 84,95 | 13,6 | 1,7-7,6 |
| Perfil autoinmune U/L | |||
| Anti-SSA (Ro) IgG U/L | 3,0 | 85,79 | Positivo mayor a 20 |
| Anti-SSB (La) IgG U/L | 3,0 | 102,91 | Positivo mayor a 20 |
| Anti-SM IgG U/L | 3,0 | 3,65 | Positivo mayor a 20 |
| Anti-RNP IgG U/L | 3,32 | 17,25 | Positivo mayor a 20 |
| ANA | Negativos | Negativos | |
| C3 mg/dL | 15 | 131 | 90-180 |
| C4 mg/dL | 25,2 | 14 | 10-40 |
ANA: anticuerpos antinucleares; anti-RNP: anticuerpos antirribonucleoproteína; anti-SM: anticuerpos anti-Smith; anti-SSA (Ro): anticuerpos anti-Ro; C3: complemento sérico C3; C4: complemento sérico C4; g/dl: gramos/decilitro; mg/dl: miligramo/decilitro; mg/l: miligramo/litro; mmol/l: milimol/litro; U/l: unidad/litro.

La resonancia muscular de miembro inferior izquierdo evidenció infiltración grasa en los haces musculares, además de atrofia muscular, especialmente en músculos bíceps femoral, semimembranoso, semitendinoso, músculo gracilis y sartorio, vasto medial, vasto lateral, vasto intermedio y parte del cuádriceps. La electromiografía reportó un patrón de afectación miopática. Se iniciaron pulsos de metilprednisolona por 5 días y más tarde pulsos con ciclofosfamida. A pesar del descenso en los niveles de enzimas musculares, no hubo una respuesta clínica.
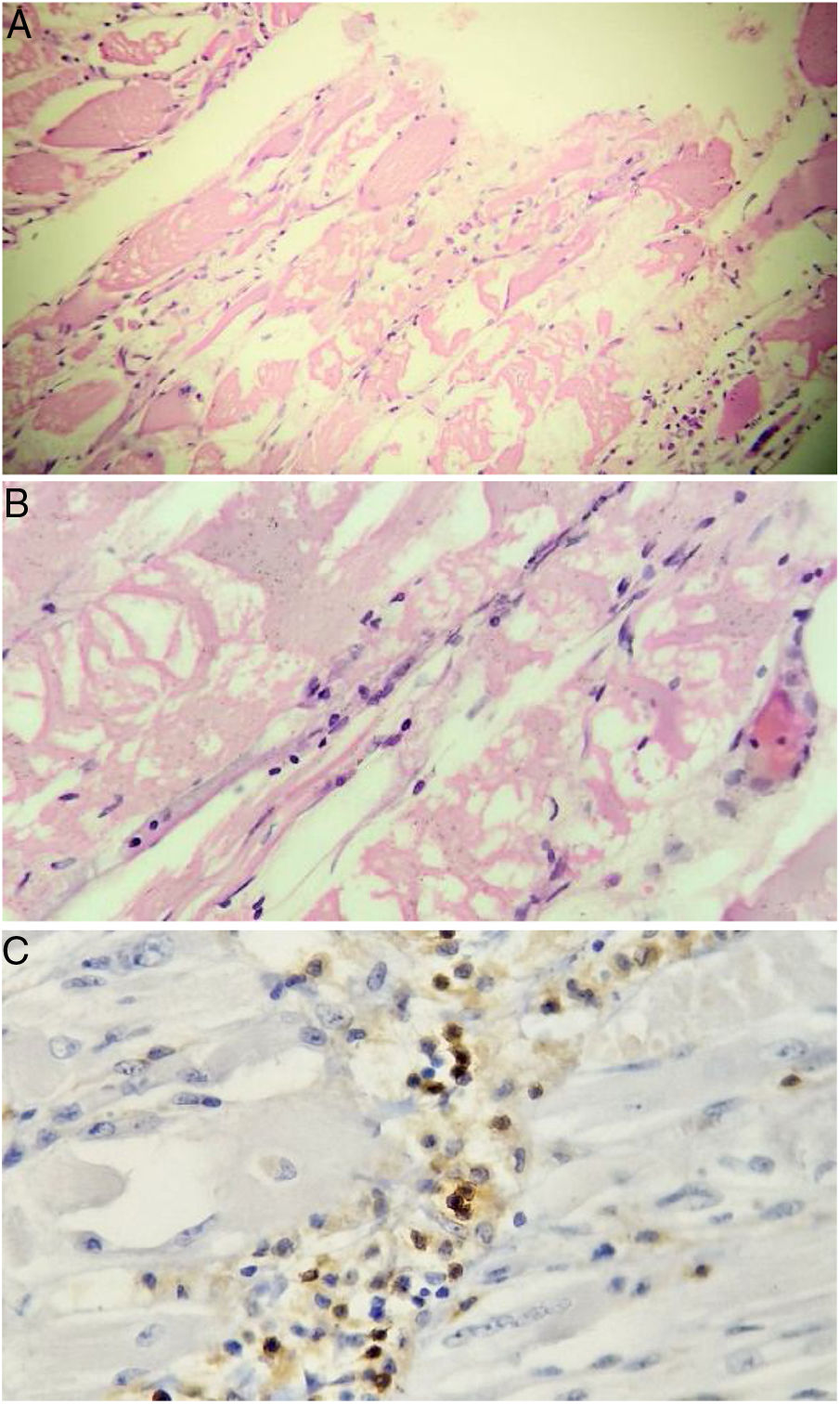
Teniendo en cuenta la pérdida de 10kg y considerando que las miopatías inflamatorias pueden asociarse a cáncer, se solicitaron estudios de extensión para descartar enfermedad neoplásica. Se realizaron tomografía axial de abdomen y pelvis, endoscopia de vías digestivas altas, ultrasonografía diagnóstica de mama, colonoscopia y ecografía de tiroides, las cuales fueron negativas para masas, adenopatías y metástasis. También se obtuvieron marcadores tumorales: ACE-CEA (antígeno carcinoembrionario), CA 125 (antígeno de cáncer de ovario), CA 19-9 (antígeno de cáncer de tubo digestivo), que también fueron negativos. Con base en el reporte de la resonancia magnética, se decidió llevar a biopsia muscular: a nivel histológico se evidenciaron cambios sugestivos de degeneración y fagocitosis miofibrilar, con importante infiltración linfocítica (fig. 2). Con lo anterior se confirmó la presencia de miositis por cuerpos de inclusión.
. Infiltrado inflamatorio linfocitario tanto endomisial como perivascular. Marcadores por inmunoperoxidasas para CD20 positivos en los infiltrados inflamatorios.")
Biopsia muscular. Se aprecian fibras musculares atróficas redondeadas de aspecto miopático de predominio perifascicular, acompañadas de cambios miopáticos (degeneración y fagocitosis miofibrilar con regeneración). Infiltrado inflamatorio linfocitario tanto endomisial como perivascular. Marcadores por inmunoperoxidasas para CD20 positivos en los infiltrados inflamatorios.
Se indicó un manejo multidisciplinario. En el curso de su evolución la paciente presentó incremento en la debilidad y disfagia, al principio para alimentos sólidos y posteriormente para líquidos; presentó, además, sialorrea y tos con la ingesta. Se solicitó una cinevideodeglución, la cual reportó alteración deglutoria grave en las fases oral y faríngea, reflujo constante y repetitivo hacia la nasofaringe, por lo que se propuso gastrostomía percutánea, que la paciente no aceptó. Luego de 30 días de hospitalización, en los cuales la paciente tuvo deterioro de su cuadro clínico, caracterizado por debilidad generalizada en el cuello y las 4 extremidades, con fuerza muscular 1/5, e incapacidad para movilización contra gravedad, no hubo respuesta exitosa a la terapia de rehabilitación física. La paciente, en compañía de su núcleo familiar, firmó salida voluntaria de la institución, a pesar de la evolución clínica y de su condición al alta.
Caso clínico 2Paciente de 61 años, diestra, con antecedente de parotiditis, síndrome de Sjögren (SS) y fenómeno de Raynaud, quien consultó por cuadro clínico de varios meses de evolución consistente en debilidad proximal generalizada progresiva de predominio en miembros inferiores con evolución lenta hasta impedir la marcha, asociado a pérdida de peso progresiva de aproximadamente 6kg. En el interrogatorio dirigido refirió síntomas sicca y fenómeno de Raynaud. Negó cambios en la piel, edema de manos, disfagia, entre otros. Sin antecedentes familiares relevantes. Al examen físico llamaba la atención la disminución de la fuerza en las 4 extremidades de predominio proximal de 4/5, sin alteración de sensibilidad ni reflejos osteotendinosos. Además, se encontró palidez distal en las manos, compatible con fenómeno de Raynaud.
Los paraclínicos iniciales se recogen en la tabla 1. Otros paraclínicos adicionales fueron: factor reumatoide 159,6 UI/mL (elevado), electroforesis de proteínas en suero con pico γpoliclonal, ecocardiograma y radiografía de tórax en parámetros normales.
Por queja, además de dolor lumbar referido, se realizó resonancia lumbosacra, en la que se evidenciaron signos de fracturas por depresión de las superficies articulares inferiores de L3 y L4 con edema de médula ósea, además de discopatía lumbar múltiple y protrusión discal que contactaba el saco dural y raíces S1. Lo anterior era sugestivo de osteoporosis y discopatía lumbar.
Se realizó electromiografía y neuroconducción de 3 extremidades, que reportó patrón de enfermedad muscular inflamatoria. La biopsia muscular documentó fibras musculares atróficas perifasciculares, con degeneración y fagocitosis miofibrilar, con presencia de vacuolas e infiltrado linfocitario perivascular compatible con MCI. Se indicó manejo con pulsos de metilprednisolona y ciclofosfamida, en adición a terapia física. En la actualidad, está en la cuarta dosis de ciclofosfamida, con evolución favorable, mejoría parcial de la fuerza en las 4 extremidades y descenso gradual de enzimas musculares.
DiscusiónLas miopatías inflamatorias son un grupo de enfermedades sistémicas que producen un cuadro clínico de debilidad muscular, elevación de enzimas musculares, hallazgos miopáticos en electromiografía y patrón inflamatorio en biopsia muscular2,3. Se clasifican de acuerdo con su presentación, inicio, manifestaciones extramusculares, características histológicas y respuesta al tratamiento. Los principales tipos son: dermatomiositis, miositis por cuerpos de inclusión, polimiositis y miopatía autoinmune necrosante1,6,7,10.
Describimos 2 casos de pacientes sin antecedentes familiares, con cuadros clínicos insidiosos de debilidad que cumplían 3 criterios de Bohan y Peter, lo que eleva la posibilidad clínica para miopatía inflamatoria; sin embargo, por alta sospecha de MCI, se aplicaron criterios de Griggs1, mediante la toma de biopsia muscular, que fueron consistentes con una miositis por cuerpos de inclusión11-13. En ambas pacientes llamó la atención la pobre respuesta al tratamiento médico instaurado y la presentación asimétrica inicial, además, en el primer caso, la rápida evolución a compromiso distal. Todas estas características se han descrito como predictores de mal pronóstico de la MCI y, además, son contrarias al curso de una polimiositis o dermatomiositis en las que, en general, se puede ver un curso clínico favorable tras el inicio del tratamiento y en las que el compromiso suele ser predominantemente proximal. Por esto, la MCI se convierte en un reto terapéutico, ya que requiere el inicio de terapia inmunomoduladora sin una efectividad óptima10.
En una serie de 30 casos de MCI publicada en Brasil, con seguimiento a 30 años14, se encontró que las principales manifestaciones, aparte del compromiso de la fuerza muscular, eran la disfagia, pérdida de peso y alteraciones cardiacas, y menos comunes eran las artralgias, síntomas respiratorios y la disfonía. En ambos casos reportados, los pacientes manifestaron pérdida de peso, sin embargo, solo el primero reportó disfagia. Ninguno de los pacientes manifestó síntomas cardiacos.
La disfagia ha sido relacionada como complicación de MCI, en la que se ha encontrado que cerca del 38% de los pacientes pueden manifestarla15. Otra complicación importante es la insuficiencia respiratoria. A pesar de una búsqueda exhaustiva, no se encontró ninguna manifestación clínica ni paraclínica de etiología neoplásica. Aunque la MCI es una causa rara de síndrome paraneoplásico, según lo expuesto por Dardis et al., es importante la búsqueda de malignidad, principalmente con escaneo tomográfico y ecografía de tiroides, debido a hallazgos post mortem de enfermedad maligna en estos pacientes16.
La correlación de MCI con SS se ha encontrado reportada en la literatura. Un caso publicado por Misterska-Skora et al.17 muestra el caso de una paciente de 57 años con síntomas secos, fenómeno de Raynaud y debilidad muscular proximal y distal de las 4 extremidades, a quien finalmente se diagnoticó de MCI y SS, con mejoría clínica posterior al inicio del tratamiento, y en quien la terapia concomitante del SS, según se concluyó, podría haber mejorado el pronóstico de la enfermedad muscular, cuadro clínico similar a lo reportado en nuestro segundo caso.
El diagnóstico de la miositis por cuerpos de inclusión puede ser difícil, dado que la presentación clínica tiene una evolución lenta y en los primeros años puede ser inespecífica. Por lo anterior, se requiere un alto índice de sospecha y hacer una asociación integral de la historia clínica, los niveles de enzimas musculares y estudios complementarios, como biopsia y electromiografía11,12. Es necesario complementar el estudio de estos pacientes para descartar coexistencia de enfermedades autoinmunes y malignidad. Los anticuerpos citoplasmáticos contra antígenos Mi-2, los anticuerpos anti-tRNA sintetasa, anti-SRP, el factor de transcripción 1-γy el gen 5 asociado a la diferenciación de melanoma son de utilidad para tal fin. En ninguno de los 2 casos presentados fue posible la toma de estos anticuerpos1,3,5,9.
Dentro de los diagnósticos diferenciales a las miopatías inflamatorias, se deben considerar las miopatías metabólicas: trastornos del almacenamiento de glucógeno (enfermedad de McArdle, enfermedad de Pompe), miopatías genéticas: distrofia muscular de la cintura escapular, facioescapulohumeral, distrofia miotónica, miopatías miofibrilares, miopatías congénitas, canalopatías del sistema musculoesquelético. Enfermedad neurológica: esclerosis lateral amiotrófica, atrofia muscular espinobulbar (enfermedad de Kennedy), polineuropatía desmielinizante inflamatoria crónica, hiperexcitabilidad del nervio periférico y radiculopatías18.
Otras enfermedades adquiridas con manifestaciones miopáticas: miositis infecciosa y posinfecciosa, endocrinopatías (hiper- o hipotiroidismo, acromegalia, síndrome de Cushing, enfermedad de Addison, deficiencia de vitamina D, hiper- o hipocalcemia, hipopotasemia), pionecrosis y traumatismos. También se deben considerar el uso de fármacos o toxinas implicados en miotoxicidad y asociados al uso de estatinas, fibratos, colchicina, hidroxicloroquina, zidovudina, cocaína, alcohol, penicilamina, entre otros18.
La biopsia muscular tiene un papel importante como parte del proceso de diagnóstico en la evaluación de un paciente con una condición neuromuscular y es esencial para la confirmación de la MCI, en la que se describen usualmente signos de cronicidad como fibras hipertróficas, atróficas, particiones, núcleos centrales e infiltración grasa. Los hallazgos histológicos «mayores» son infiltrado linfocítico multifocal que invade fibras no necróticas, vacuolas en células no invadidas por linfocitos (estas vacuolas tipo rimmed contienen depósitos granulares basófilos) y depósito de amiloiderojo del Congo positivo. Es frecuente el hallazgo de fibras del tipo ragged red o «rojas rasgadas» y citocromo-oxidasa negativas, como consecuencia de disfunción mitocondrial5,9,19. En ambas biopsias reportadas de las pacientes fue consistente el infiltrado linfocítico multifocal y la fagocitosis miofibrilar con presencia de vacuolas, compatible con lo descrito en la literatura para esta entidad.
El pronóstico de la enfermedad es ominoso, teniendo en cuenta el curso progresivo e incapacitante. Se pone énfasis en que estos pacientes requieren un manejo interdisciplinario que incluya la participación de neurología, medicina interna, psiquiatría, psicología, fisiatría, rehabilitación física y terapia respiratoria, entre otros10.
ConclusiónLa miositis por cuerpos de inclusión es un diagnóstico que considerar en pacientes con debilidad asimétrica tanto proximal como distal, con títulos bajos de creatinina cinasa y en aquellos que no responden a terapia convencional. Es importante alcanzar un diagnóstico temprano con el fin de evitar complicaciones que pudieran empeorar su pronóstico. El tratamiento debe incluir terapia inmunosupresora, acompañamiento psicológico y una rehabilitación física óptima con el fin de impactar la calidad de vida de los pacientes.
FinanciaciónNinguna.
Conflicto de interesesNo hay conflicto de intereses.
