




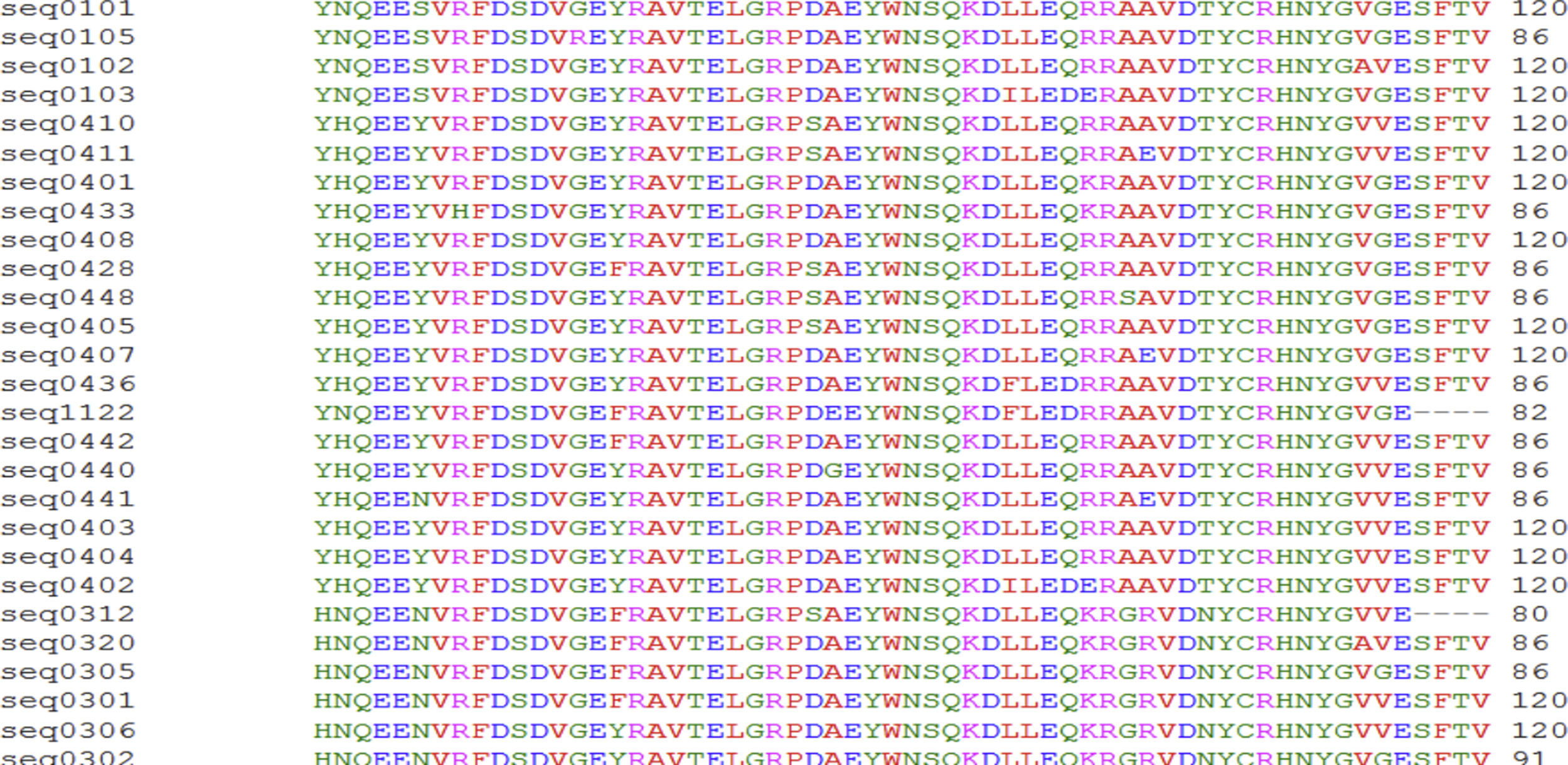




Utilizar modelos de inteligencia computacional para la clasificación e identificación de endofenotipos (relación entre fenotipo y marcadores genéticos) en pacientes con artritis reumatoide y controles sanos, a partir de información genética, principalmente el HLA DRB1 (antígeno leucocitario humano) y la teoría del epítope compartido. Esto hace referencia a la asociación entre la artritis reumatoide y el HLA DRB1, principalmente los alelos que contienen un motivo común de aminoácidos de las secuencias QKRAA, QRRAA o RRRAA en las posiciones 70 a 74 de la cadena del DRB1, los cuales confieren una susceptibilidad particular de la enfermedad a los individuos.
MétodosDesarrollamos modelos computacionales para clasificación, utilizando técnicas de inteligencia computacional como son las redes neuronales, redes bayesianas y métodos como k-means. Como datos de entrada se utilizaron variables como: factor reumatoide, anticuerpos contra péptido citrulinado, proteína C reactiva, número de articulaciones inflamadas y dolorosas, rigidez matinal, edad, género, antecedentes de comorbilidades y la información del alelo HLA DRB1. Se utilizaron técnicas de bioinformática para la búsqueda de secuencias de aminoácidos relacionadas con la severidad de la enfermedad.
ResultadosSe obtuvieron resultados importantes para el diagnóstico de la enfermedad, así como también para su categorización y como potencial aplicación en la medicina personalizada de los individuos afectados por esta enfermedad. Se diseñaron modelos computacionales para clasificación, dentro de los cuales la red neuronal utilizando 5 variables obtuvo una sensibilidad del 92,3% con una especificidad del 86,66% y la red bayesiana logró una sensibilidad del 92,3% y una especificidad del 93,33%. La red neuronal utilizando 11 variables obtuvo una sensibilidad del 84,61% y una especificidad del 93,33% mientras que la red bayesiana consiguió una sensibilidad del 92,3% con una especificidad del 93,33%. Un método de agrupamiento tipo k-means fue utilizado para dividir en 2 grupos la totalidad de pacientes y controles. Además, se logró separar 2 grupos entre los pacientes para definir su severidad. Finalmente, se consiguió obtener un árbol de distancia entre secuencias de aminoácidos de los distintos alelos HLA DRB1, lo cual permite visualizar grupos con cercanía genética y lograr determinar que posiblemente existen más grupos fuera de los propuestos en las distintas teorías.
ConclusiónLos métodos utilizados permiten una mejor estratificación de la enfermedad en relación con la predicción de fenotipos y posibles desenlaces de la enfermedad, así como para la potencial prevención primaria de la enfermedad.
To use computational intelligence models for the classification and identification of endophenotype (relationships between phenotype and genetic markers) in patients with rheumatoid arthritis and healthy controls from genetic information, primarily the DRB1 HLA (human leukocyte antigen) and the shared epitope theory. This refers to the association between rheumatoid arthritis and the HLA-DRB1 alleles mainly containing amino acid common motif sequences QKRAA, RRRAA, QRRAA or at positions 70 to 74 DRB1 chain, which have been associated to susceptibility of this disease.
MethodsComputational models were developed for classification using computational intelligence techniques, such as neural networks, Bayesian networks, and methods such as k-means. The input data consisted of variables such as: rheumatoid factor, anti-citrullinated protein antibody, C-reactive protein, number of swollen and tender joints, morning stiffness, age, gender, history of comorbidities, and the information on the HLA-DRB1. Bioinformatics techniques were used to search for amino acid sequences related to disease severity.
ResultsPromising results for the diagnosis of the disease were obtained, as well as its categorisation as potential application in personalised medicine for individuals suffering from this disease. Computational models were designed for the classification, in which the neural network using 5 variables obtained a sensitivity of 92.3% with a specificity of 86.66%, and the Bayesian network obtained a sensitivity of 92.3% and a specificity of 93.33%. The neural network using 11 variables had a sensitivity of 84.61% and a specificity of 93.33%, while the Bayesian network obtained a sensitivity of 92.3% with a specificity of 93.33%. K-means clustering method type was used to divide all patients and controls into two groups of data. It also managed to obtain two patient groups to define severity. Finally, a tree distance was obtained between amino acid sequences of the different alleles HLA DRB1, which allows genetic proximity groups to be visualised and to determine and ensure that there are may be more groups outside the proposed different theories.
ConclusionThe proposed method can be used to provide better stratification of the disease in relation to the predicted phenotypes, and the potential for primary prevention of this disease.
La artritis reumatoide (AR) es una enfermedad autoinmune que afecta al 0,5-1% de la población humana. Existen algunos marcadores genéticos implicados en esta enfermedad1. Está asociada con discapacidad progresiva, complicaciones sistémicas, muerte temprana y altos costos socioeconómicos. La causa es desconocida y su pronóstico es reservado2.
El más importante factor de riesgo genético para la AR se encuentra en el HLA. En particular los que contengan las secuencias de aminoácidos QKRAA, QRRAA o RRRAA en las posiciones 70-74 de la cadena DRB1. Esto es conocido como epítope compartido (EC)1,2.
El curso clínico de la enfermedad difiere de un individuo a otro y los factores genéticos ligados a HLA DR han surgido como potencial factor pronóstico de esta enfermedad. El EC ha sido asociado con la predisposición y progresión de la enfermedad3,4.
Los criterios para el diagnóstico de AR están definidos por el Colegio Americano de Reumatología y el EULAR 20105. Existe predominio en el género femenino con una relación mujer hombre de 3:1, la herencia de la AR es poligénica y no sigue un patrón mendeliano. La importancia de encontrar factores genéticos asociados con la AR radica en la contribución a la comprensión de los mecanismos patogénicos de la enfermedad y su posible aplicación clínica para la identificación de marcadores, diagnóstico y pronóstico.
Modelos computacionales, como las redes neuronales artificiales, son sistemas basados en la computación paralela masiva que permiten realizar tareas de control y de clasificación. Una red neural biológica puede ser modelada matemáticamente por un grafo dirigido con nodos (neuronas) interconectados. Los nodos artificiales son casi siempre funciones simples cuyos argumentos son las sumas de las entradas al nodo; algunos trabajos sobre redes neuronales utilizan funciones de nodo que manejan solo valores binarios6,7.
Los enfoques bayesianos proporcionan una forma consistente para hacer inferencia, mediante la integración de la evidencia de los datos con conocimientos previos del problema. Una red bayesiana puede superar la principal dificultad de controlar la complejidad del modelo en la construcción de una red neuronal estándar. Un enfoque bayesiano ofrece herramientas eficaces para evitar el sobreajuste, incluso con modelos muy complejos y, además, facilita la estimación de los intervalos de confianza de los resultados8.
Los problemas de agrupamiento surgen en muchas aplicaciones diferentes, como la minería de datos y descubrimiento de conocimiento, compresión de datos, reconocimiento de patrones y clasificación de patrones. La noción de lo que constituye un buen grupo depende de la aplicación y hay muchos métodos para encontrar grupos como k-means9.
El coeficiente silueta es utilizado para representar las particiones obtenidas por diferentes técnicas; cada grupo es representado por un coeficiente llamado silueta, el cual se basa en la comparación de cercanía y separación entre los elementos de los grupos, lo cual muestra qué objetos se encuentran correctamente dentro de un grupo y cuáles están simplemente en algún lugar entre las agrupaciones. Esta combinación de los coeficientes silueta permite una apreciación de la calidad de los grupos y una visión general de la configuración de los datos. Esta información proporciona una evaluación de la agrupación y podría ser utilizada para seleccionar un número adecuado de agrupaciones10,11. El coeficiente silueta es una medida de un enfoque que compara las distancias entre los distintos grupos formados12.
Para el análisis de los datos se usó la herramienta de software WEKA, un software de código abierto. Este sistema fue desarrollado en la Universidad de Waikato en Nueva Zelanda. WEKA representa un entorno para análisis de datos y está escrito en el lenguaje orientado por objetos Java. WEKA proporciona implementaciones de avanzados algoritmos de minería de datos y algoritmos de aprendizaje de máquina. Además, contiene módulos de preprocesamiento de datos, clasificación, agrupamiento y extracción de reglas de asociación13.
MetodologíaEn la selección del conjunto de datos utilizados por los modelos computacionales se obtuvo información de pacientes con AR y controles sanos, con previo consentimiento informado por parte de los pacientes. Al inicio del estudio se contaba con 135 pacientes y 140 controles sanos, para un total de 275 registros. Al realizar el preprocesamiento de los datos, varios de los registros fueron eliminados por presentar información faltante o inconsistente. Finalmente, se logró tener una muestra completa con 138 datos que correspondían a 63 pacientes y 75 controles sanos (tabla 1).
Los datos utilizados en este estudio corresponden a información clínica, serológica y genética de 138 personas, entre pacientes con AR y controles sanos. Su distribución se puede observar en la tabla 2.
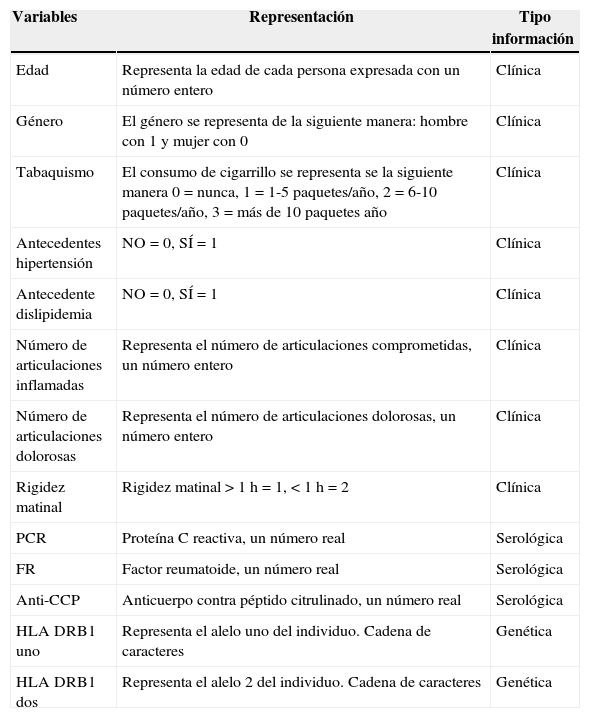
Variables utilizadas dentro del estudio
| Variables | Representación | Tipo información |
|---|---|---|
| Edad | Representa la edad de cada persona expresada con un número entero | Clínica |
| Género | El género se representa de la siguiente manera: hombre con 1 y mujer con 0 | Clínica |
| Tabaquismo | El consumo de cigarrillo se representa se la siguiente manera 0=nunca, 1=1-5 paquetes/año, 2=6-10 paquetes/año, 3=más de 10 paquetes año | Clínica |
| Antecedentes hipertensión | NO=0, SÍ=1 | Clínica |
| Antecedente dislipidemia | NO=0, SÍ=1 | Clínica |
| Número de articulaciones inflamadas | Representa el número de articulaciones comprometidas, un número entero | Clínica |
| Número de articulaciones dolorosas | Representa el número de articulaciones dolorosas, un número entero | Clínica |
| Rigidez matinal | Rigidez matinal>1h=1, <1h=2 | Clínica |
| PCR | Proteína C reactiva, un número real | Serológica |
| FR | Factor reumatoide, un número real | Serológica |
| Anti-CCP | Anticuerpo contra péptido citrulinado, un número real | Serológica |
| HLA DRB1 uno | Representa el alelo uno del individuo. Cadena de caracteres | Genética |
| HLA DRB1 dos | Representa el alelo 2 del individuo. Cadena de caracteres | Genética |
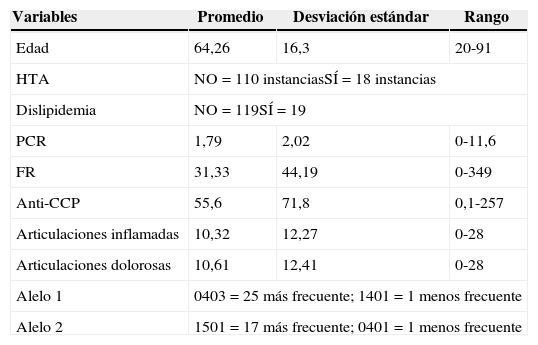
Después del preprocesamiento de los datos se realizó el análisis estadístico de las variables cuyos resultados se presentan en la tabla 3.
Análisis estadísticos de los datos
| Variables | Promedio | Desviación estándar | Rango |
|---|---|---|---|
| Edad | 64,26 | 16,3 | 20-91 |
| HTA | NO=110 instanciasSÍ=18 instancias | ||
| Dislipidemia | NO=119SÍ=19 | ||
| PCR | 1,79 | 2,02 | 0-11,6 |
| FR | 31,33 | 44,19 | 0-349 |
| Anti-CCP | 55,6 | 71,8 | 0,1-257 |
| Articulaciones inflamadas | 10,32 | 12,27 | 0-28 |
| Articulaciones dolorosas | 10,61 | 12,41 | 0-28 |
| Alelo 1 | 0403=25 más frecuente; 1401=1 menos frecuente | ||
| Alelo 2 | 1501=17 más frecuente; 0401=1 menos frecuente | ||
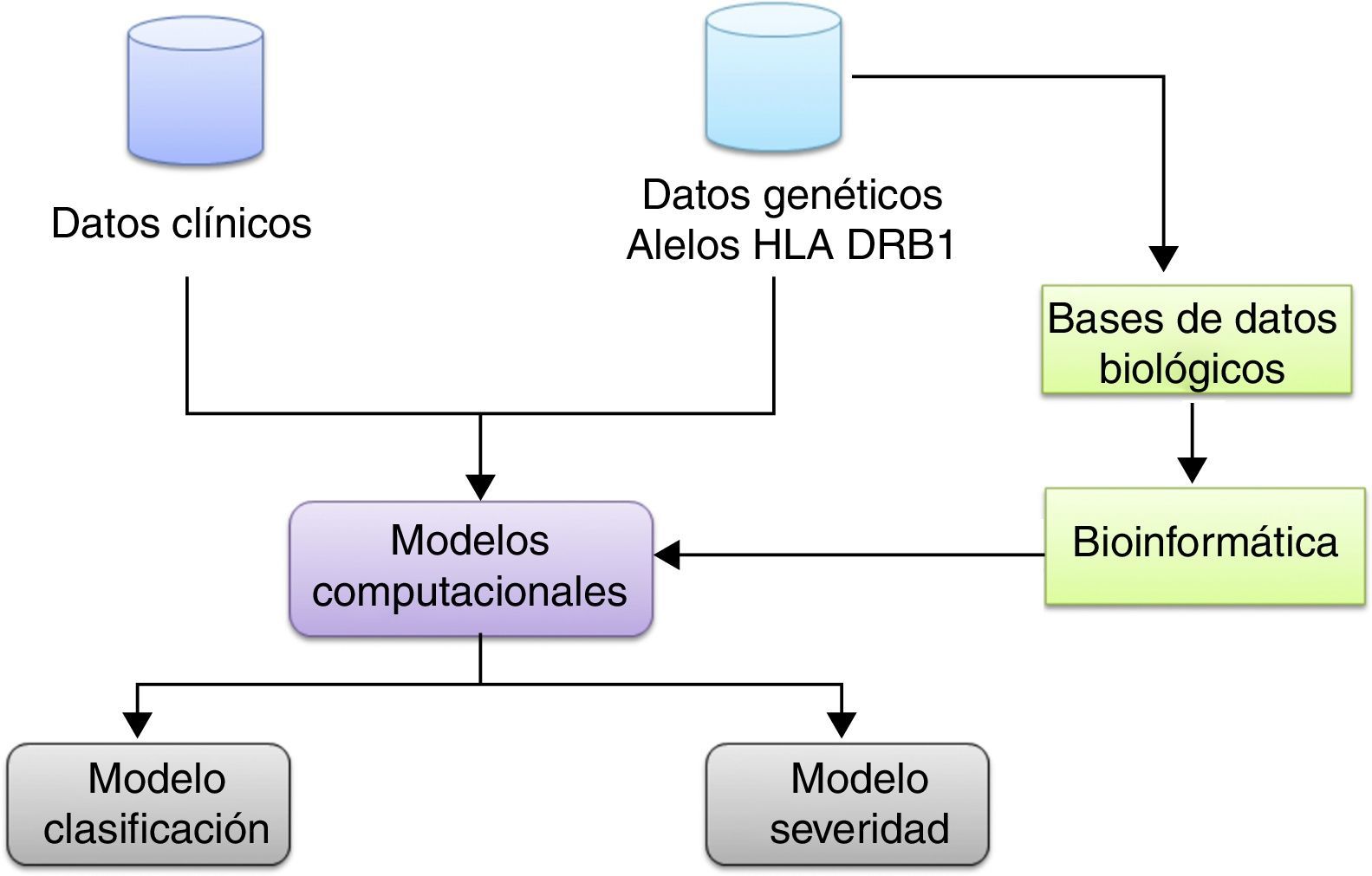
La metodología general utilizada en el trabajo se representa en el esquema de la figura 1.

Se realizó la búsqueda y selección de la información clínica y genética, principalmente el HLA DRB1 de los pacientes y controles. De los 138 registros, que corresponden a 63 pacientes y 75 controles, se seleccionó el 80% de los datos para entrenamiento y el 20% de los datos para prueba de manera estratificada (tabla 3).
Para el proceso de búsqueda de las secuencias de aminoácidos para cada uno de los alelos correspondientes a los datos, se realizó la búsqueda de los aminoácidos correspondientes a cada alelo en bases de datos biológicos que contienen esta información. Se utilizó, principalmente, la base de datos IMGT/HLA (ImMunoGeneTics Project). Posteriormente, se realizó un alineamiento entre las secuencias obtenidas utilizando una matriz tipo Blosum. Finalmente, se aplicaron métodos computacionales para la clasificación y técnicas de bioinformática, para la identificación de endofenotipos.
Para el modelamiento e implementación de los modelos computacionales basados en técnicas de inteligencia computacional, como redes neuronales tipo perceptrón, perceptrón multicapa, redes bayesianas y métodos de agrupamiento como k-means, se utilizaron herramientas computacionales como WEKA, Rapidminer y Jalview.
WEKA y Rapidminer se usaron para implementar las redes neuronales, las redes bayesianas y para realizar los procesos de agrupación. Por otro lado, se utilizó Jalview para analizar las secuencias de aminoácidos. Jalview permite realizar alineamientos múltiples, editar los alineamientos y realizar estudios de cercanía genética para efectuar estudios específicos.
Para la agrupación de los datos entre pacientes y controles se utilizó k-means y como criterio de selección del número de grupos se utilizó el coeficiente de silueta.
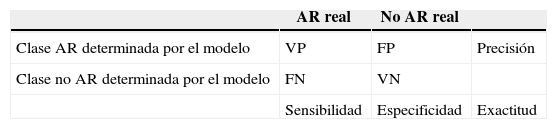
ResultadosLos resultados obtenidos se evaluaron usando las medidas de desempeño como: sensibilidad, especificidad, precisión y exactitud, que son definidas con base en verdaderos positivos (VP), verdaderos negativos (VN), falsos positivos (FP), falsos negativos (FN), número de muestras positivas (P) y número de muestras negativas (N). Las ecuaciones correspondientes se presentan a continuación:
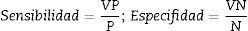
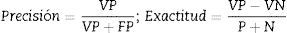
La matriz de confusión que se utilizó para presentar los resultados de las medidas de desempeño de cada modelo se presenta en la tabla 4.
El primer escenario que se plantea es utilizar los criterios del Colegio Americano de Reumatología para clasificación de la AR, para lo cual se usaron las 5 variables implicadas en estos criterios: articulaciones inflamadas, articulaciones dolorosas, factor reumatoide (FR), proteína C reactiva (PCR) y anticuerpos contra péptido citrulinado (anti-CCP), entre otras.
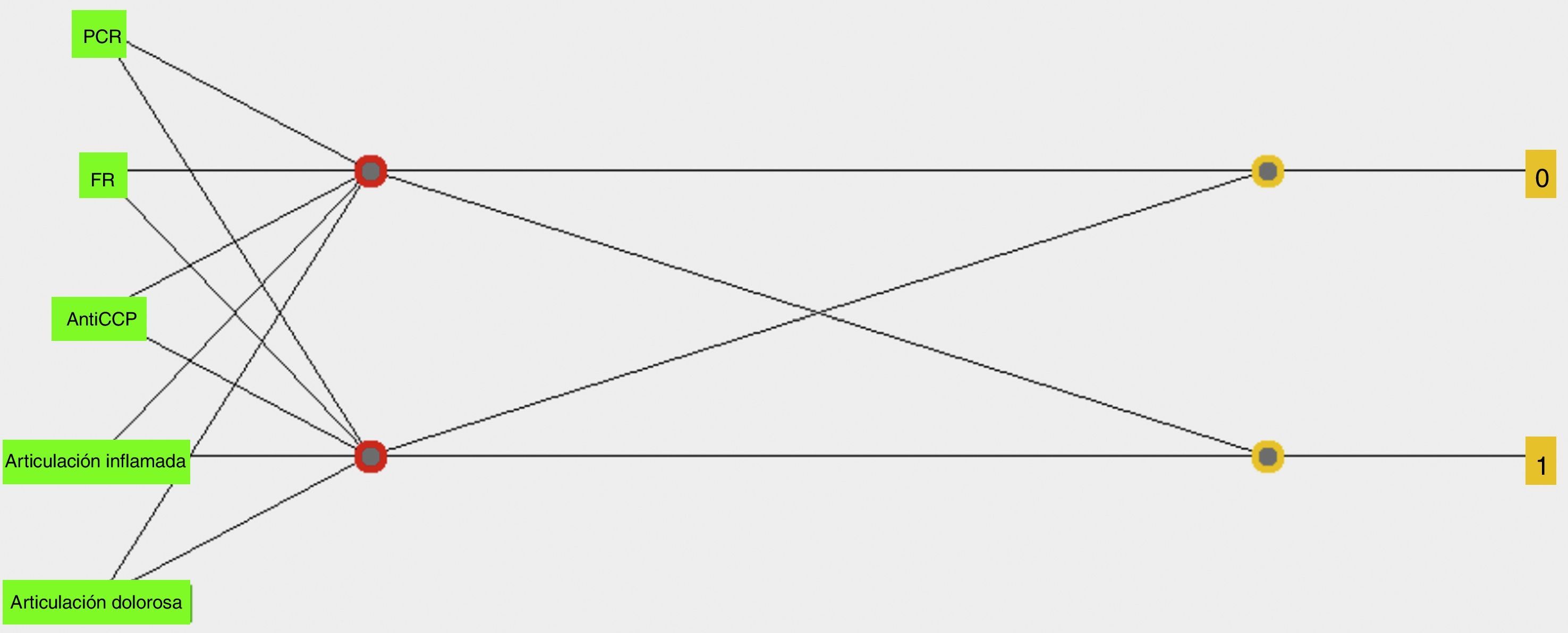
Como preprocesamiento de los datos se normalizaron todas las variables, se utilizó una red neuronal tipo perceptrón multicapa con una capa oculta que contiene 7 neuronas, se optimizaron parámetros, se utilizó el 80% de los datos para entrenamiento y el 20% para prueba. En la figura 2 se puede observar la red neuronal que se obtuvo.

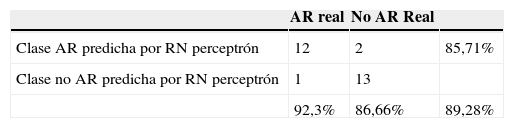
La matriz de confusión y las medidas de desempeño utilizando la red neuronal con 5 variables se presentan en la tabla 5, la cual muestra que el modelo presenta una exactitud del 89,28%, una precisión del 85,71%, con una sensibilidad del 92,3% y una especificidad del 86,66%.
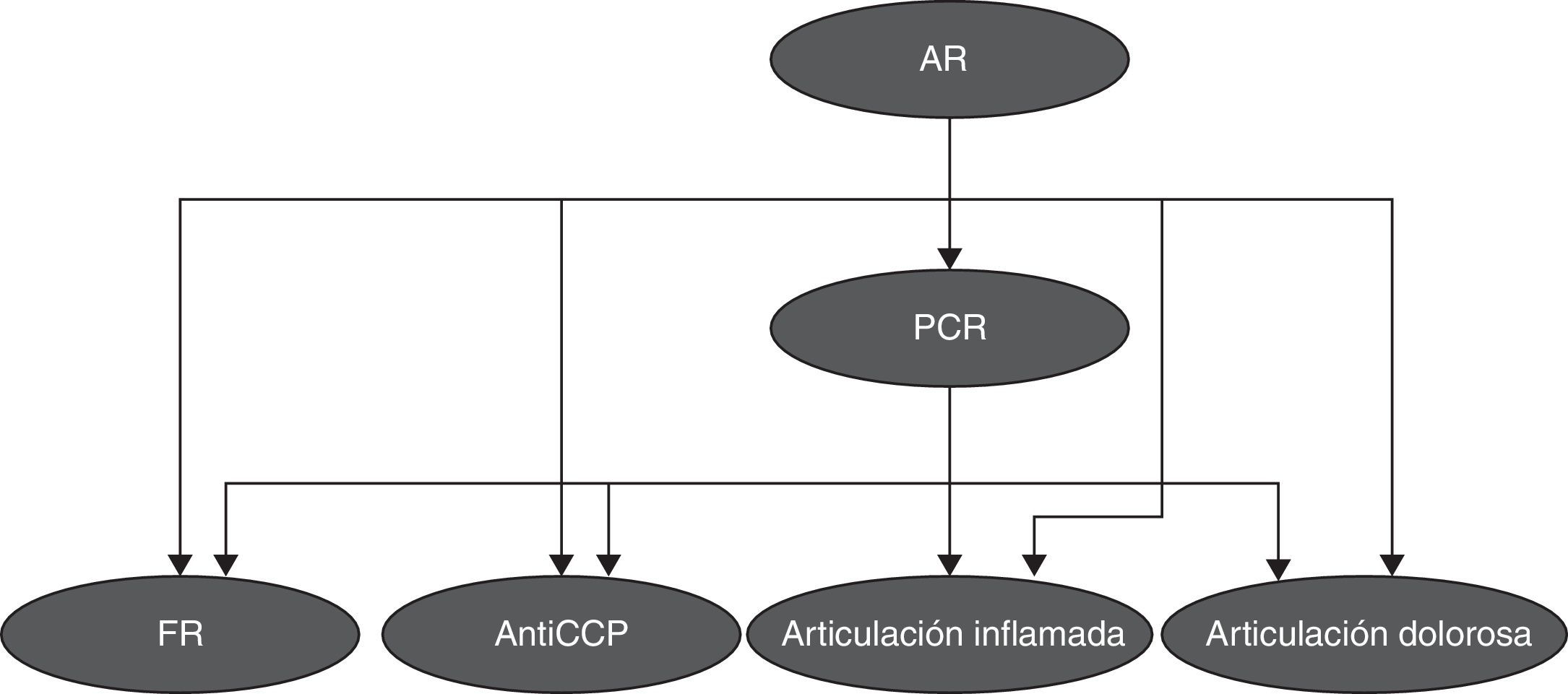
Después se implementó una red bayesiana utilizando las 5 variables propuestas por el Colegio Americano de Reumatología, la red bayesiana fue desarrollada utilizando un estimador simple y un algoritmo de búsqueda TAN (Tree Augmented Naive Bayes), la red obtenida se muestra en la figura 3.

El diseño de la red bayesiana muestra la relación de la variable PCR con las otras variables FR, anti-CCP, articulaciones dolorosas y articulaciones inflamadas. De esta manera, la probabilidad de determinar AR o no AR se da a partir de la tabla de probabilidad de PCR, la cual está directamente relacionada con las otras variables.
La matriz de confusión y los valores de las medidas de desempeño de la red bayesiana utilizando 5 variables se pueden observar en la tabla 6.
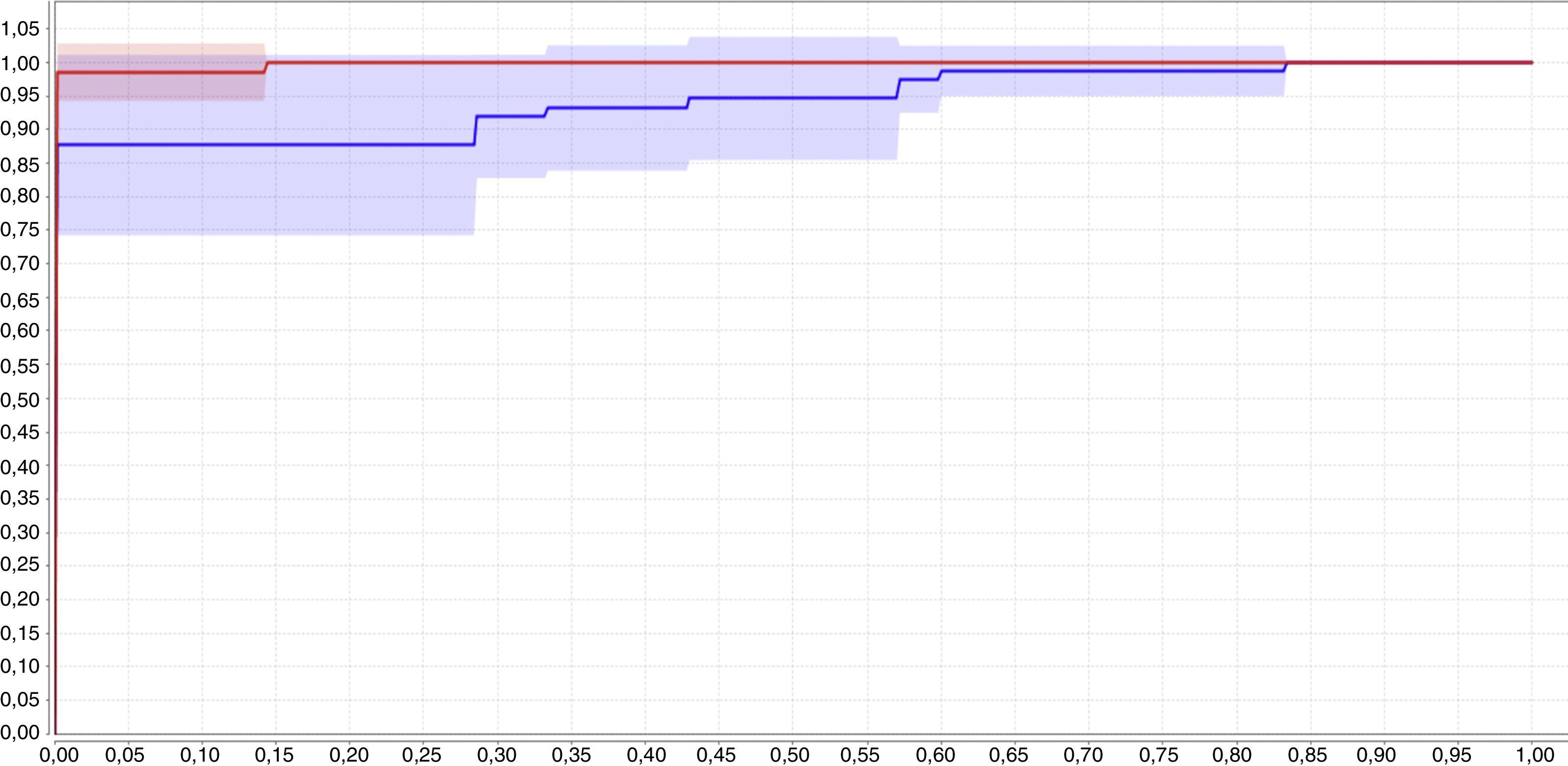
En la figura 4 se muestran las curvas ROC para los 2 clasificadores de red neuronal y red bayesiana utilizando 2 variables, el color rojo corresponde a la red bayesiana tipo perceptrón multicapa y la curva azul corresponde a la red neuronal. En la figura 4 se observa que, según la curva ROC, el clasificador bayesiano, representado en color rojo, tiene un mejor desempeño que el clasificador neuronal, representado en color azul.

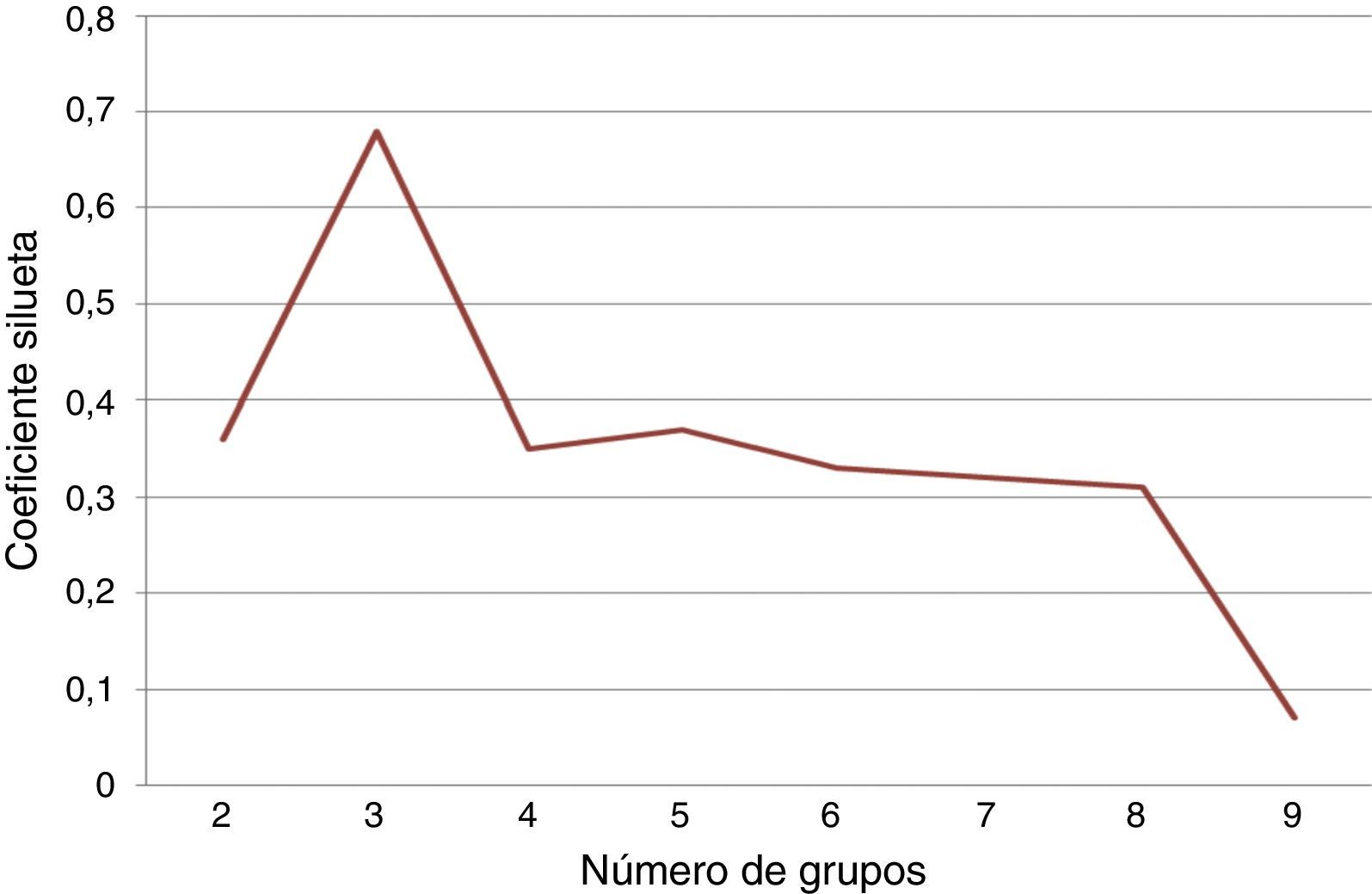
También se utilizó k-means como método de agrupación para los datos, considerando 5 variables según los criterios del Colegio Americano de Reumatología: PCR, FR, anti CCP, número de articulaciones dolorosas y número de articulaciones inflamadas. Se utilizó el coeficiente silueta para determinar el mejor k utilizando una distancia euclidiana. En la figura 5 se muestra la gráfica que presenta el coeficiente silueta con respecto a cada k posible, en la cual se comprueba que el mejor coeficiente se obtiene con un k igual a 2.

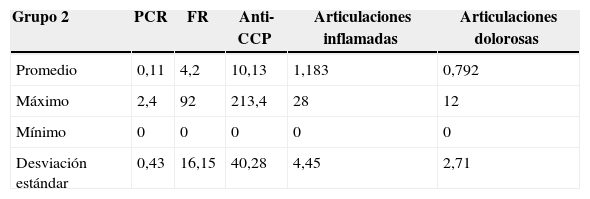
A partir de los 138 datos se realizó el agrupamiento utilizando k-means sobre las variables normalizadas. Los 2 grupos se conformaron de la siguiente manera: el primer grupo con 56 instancias y el segundo con 82 instancias. El análisis estadístico de los 2 grupos obtenidos se puede observar en las tablas 7 y 8.
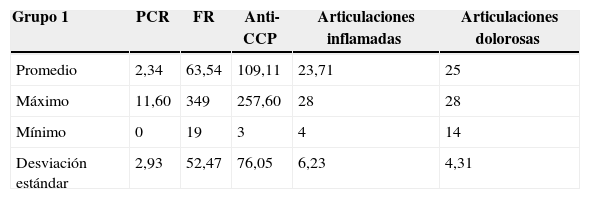
En el grupo 1 se observa que el valor del promedio del FR es de 63,54, el cual es un valor positivo con respecto a los valores normales para dicho examen de laboratorio y, además, es mayor al obtenido en el grupo 2 que fue de 4,2. La desviación estándar del anti-CCP del grupo 1 es de 76,05 con respecto a 40,28 del grupo 2, con lo cual se deduce que los grupos no son homogéneos como se esperaba.
Los promedios del número de articulaciones inflamadas y dolorosas del grupo 1 fueron de 23,71 y 25, respectivamente. En contraste, los promedios del grupo 2 fueron de 1,183 y 0,792. Lo anterior indica que los individuos del grupo 1 tienen un compromiso articular mayor con respecto al grupo 2.
En el grupo 2 el promedio de los datos para la variable PCR fue de 0,11, el cual corresponde a un valor cualitativo negativo para dicho examen de laboratorio, siendo el máximo de 2,4 en este grupo.
Por otro lado, en el grupo 1 el promedio fue de 2,34 con un valor máximo de 11,6.
Los valores promedio de anti-CCP varían considerablemente en los 2 grupos, siendo el promedio del grupo 1 de 109,11 y el del grupo 2 de 10,13. Lo que indica, según lo reportado en la literatura, una predisposición mayor a presentar la enfermedad. Al examinar estos resultados se puede afirmar que los individuos del grupo 1 presentan una actividad mayor de la enfermedad con respecto a los individuos del grupo 2.
Los estudios han demostrado que una característica de la AR es la presencia de autoanticuerpos. En la enfermedad el FR puede detectarse con una sensibilidad del 60-70% y una especificidad del 80-90%. Sin embargo, ensayos de detección de anticuerpos anti-CCP tienen una mayor especificidad (98%), con una sensibilidad del 68-80%. Estudios retrospectivos, realizados en diferentes países, han demostrado que los autoanticuerpos, incluyendo anti-CCP, anticuerpos contra antígenos que contiene el aminoácido citrulina y FR, pueden ser detectados en pacientes varios años antes de que los síntomas clínicos de la AR se presenten y que están asociados directamente con la enfermedad, por lo tanto, la presencia de anticuerpos anti-CCP son un factor de riesgo para presentar AR14.
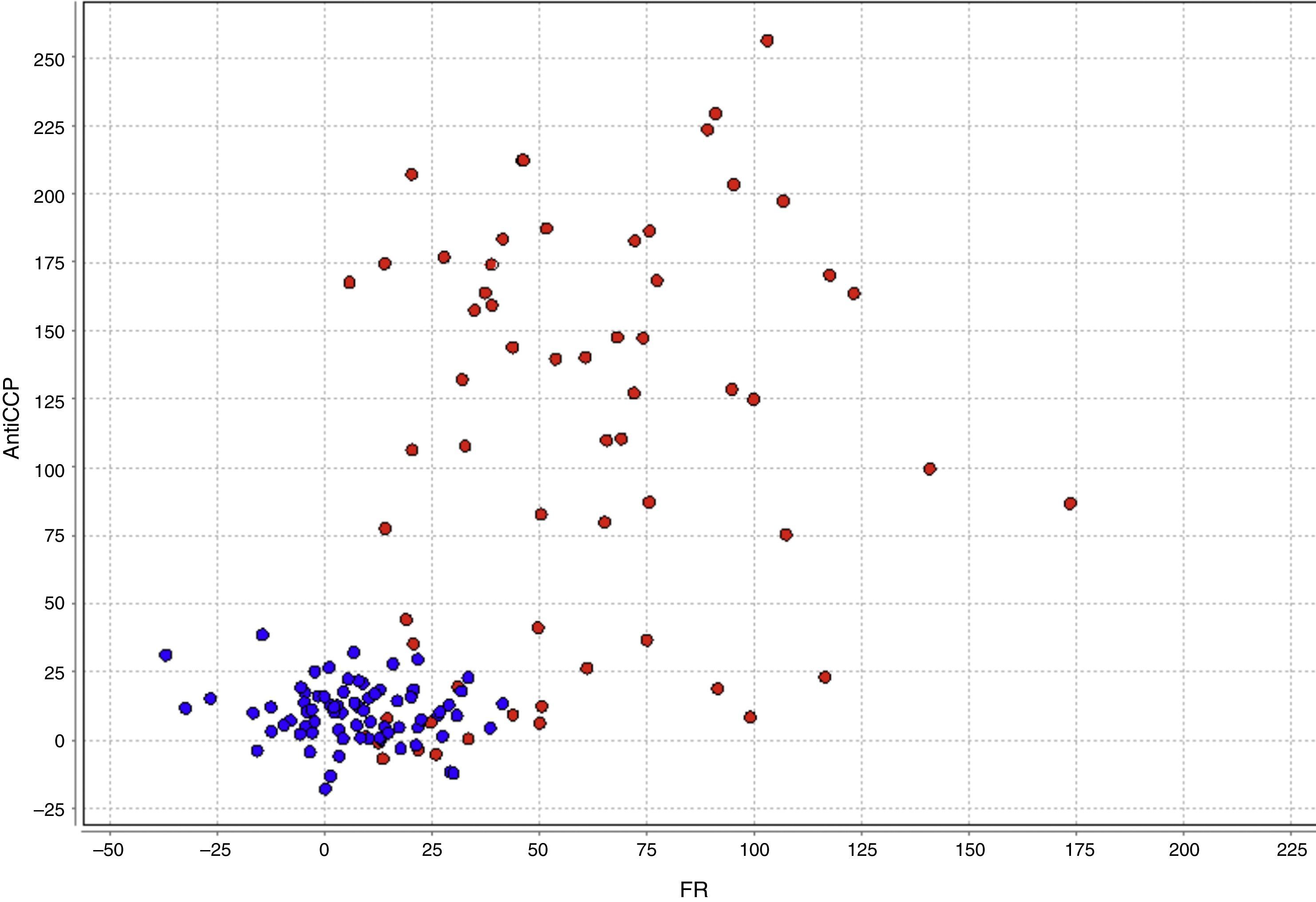
En la figura 6 se grafican las variables FR y anti CCP representándose con color rojo el grupo 1 y con color azul el grupo 2. En el gráfico se puede observar que los individuos del grupo 1 tienen valores mayores en la variables anti-CCP con respecto a los de los individuos del grupo 2, lo cual confirma los resultados reportados previamente en la literatura.

También se diseñaron e implementaron modelos computacionales utilizando 11 variables: 2 variables genéticas correspondientes a los 2 alelos del HLA DRB1; las variables serológicas PCR, FR y anticuerpos anti-CCP y 6 variables clínicas: edad, antecedente de tabaquismo, antecedente de hipertensión arterial, antecedente de dislipidemia, número de articulaciones dolorosas y número de articulaciones inflamadas.
La red neuronal diseñada tuvo 7 neuronas para una capa oculta. Se aplicó filtro de nominal a binario para las variables de entrada, con una tasa de aprendizaje de 0,3 y momento 0,2. La red neuronal se entrenó a través del método de retropropagación. Por otro lado, para la red bayesiana se utilizó un estimador simple y el algoritmo de búsqueda tipo TAN. Finalmente, se realizó agrupación con k-means usando una distancia euclidiana y para calcular el mejor k se utilizó el coeficiente de silueta.
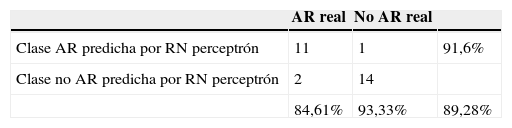
La red neuronal tipo perceptrón multicapa utilizando 11 variables obtuvo una exactitud de 89,28% que corresponde a 25 instancias correctamente clasificadas de 28 instancias de prueba. La correspondiente matriz de confusión y las medidas de desempeño se presentan en la tabla 9.
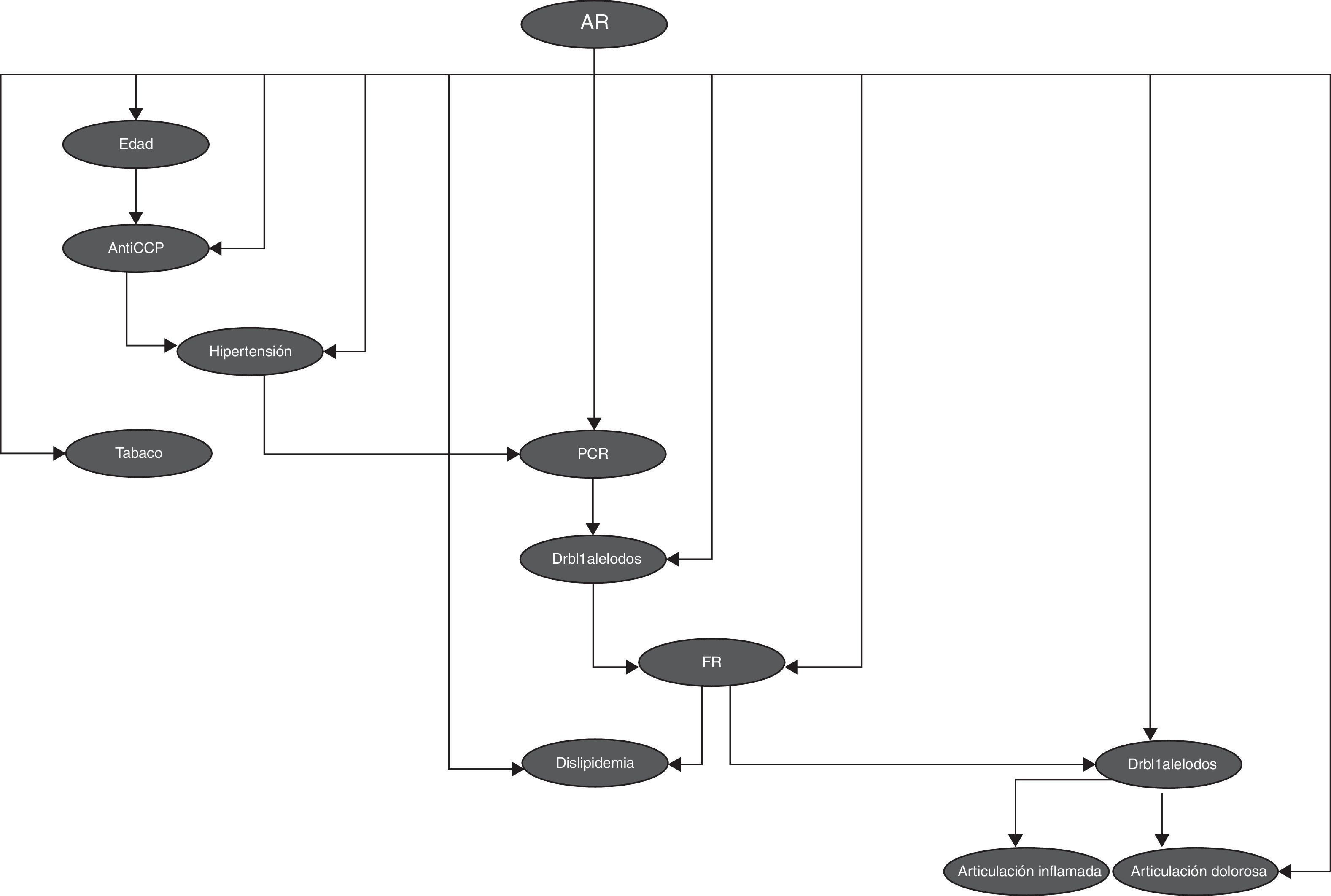
En la figura 7 se observa la red bayesiana implementada utilizando 11 variables para clasificación entre pacientes y controles, en la cual se aprecian las dependencias probabilísticas entre las variables utilizadas.

En la red bayesiana observamos las relaciones que se presentan entre las distintas variables genéticas, clínicas y serológicas, principalmente la dependencia que se ve entre la variable alelo 1 y las variables clínicas articulaciones inflamadas y articulaciones dolorosas; esto nos demuestra la relación existente entre la genética de la enfermedad y sus manifestaciones clínicas.
Con la red bayesiana se obtuvieron los siguientes resultados: de las 28 instancias de prueba se clasificaron adecuadamente 25, que corresponde al 92,85%, e incorrectamente 3 instancias, que corresponde al 7,15%, con una sensibilidad del 92,3% y una especificidad del 93,33%. La matriz de confusión y las medidas de desempeño se presentan en la tabla 10.
Los nodos en la red bayesiana se relacionan mediante probabilidades de que un suceso ocurra.
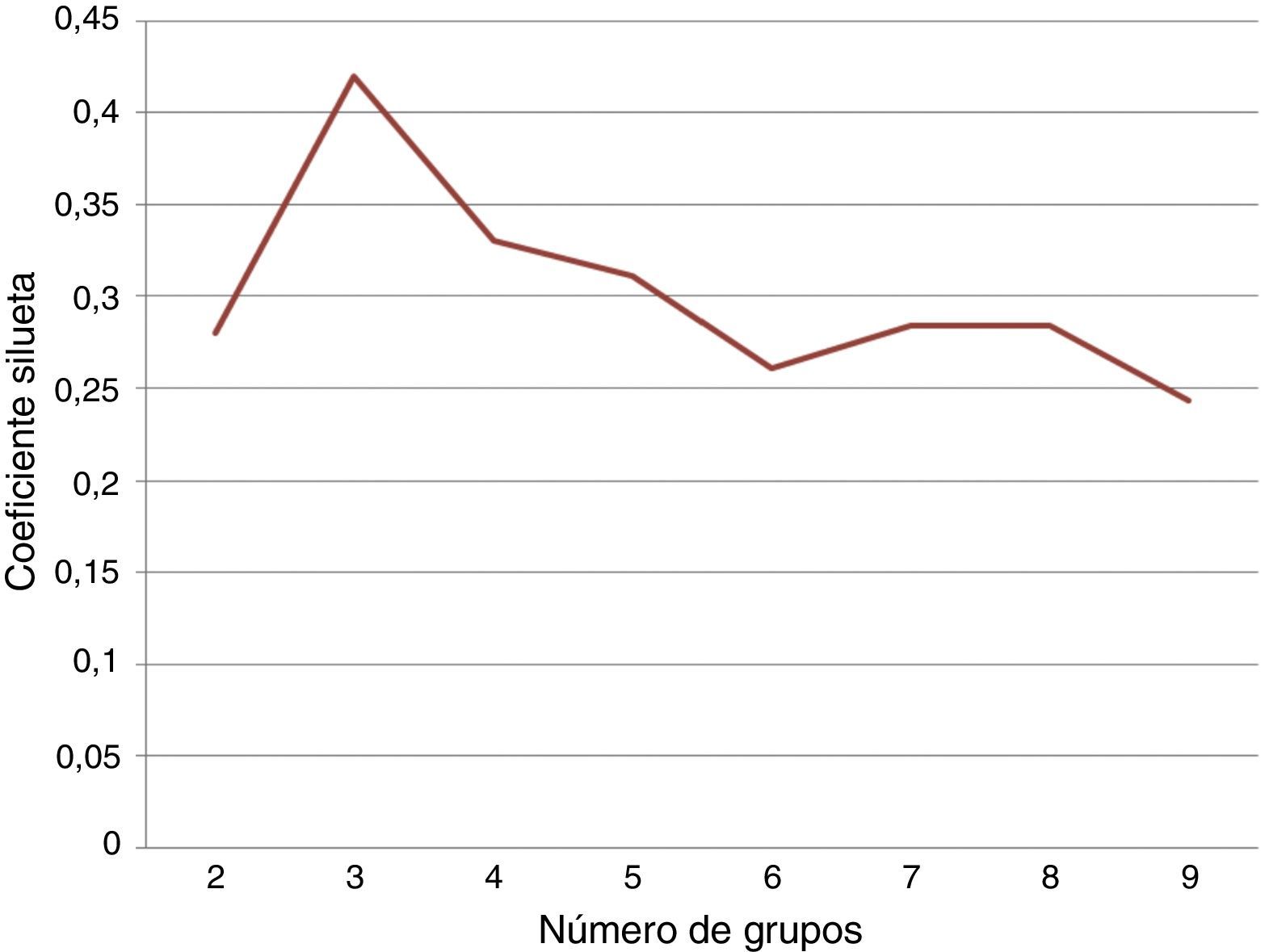
También se realizó una agrupación con los datos de los 63 pacientes. Esto forma parte del estudio de clasificación de severidad de la enfermedad. Se utilizó el valor del coeficiente silueta para determinar el mejor número de grupos. La figura 8 muestra los valores del coeficiente silueta para varios valores de k. El mejor valor del coeficiente silueta se obtiene para k=2.

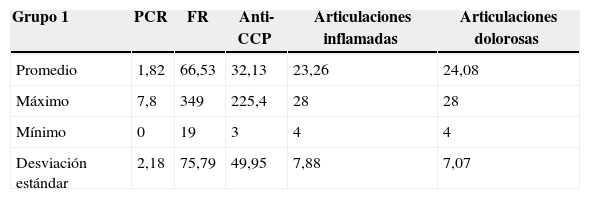
Para esto se utilizó k-means sobre las variables normalizadas para identificar entre los pacientes los aspectos relacionados con la severidad de la enfermedad. Se hizo una agrupación en 2 grupos, los cuales quedaron conformados de la siguiente manera: el primer grupo con 40 individuos y el segundo con 23. El análisis estadístico de los 2 grupos obtenidos se puede observar en las tablas 11 y 12.
Análisis estadístico de los datos que conforman el grupo 1 de pacientes teniendo en cuenta la severidad de la enfermedad
| Grupo 1 | PCR | FR | Anti-CCP | Articulaciones inflamadas | Articulaciones dolorosas |
|---|---|---|---|---|---|
| Promedio | 1,82 | 66,53 | 32,13 | 23,26 | 24,08 |
| Máximo | 7,8 | 349 | 225,4 | 28 | 28 |
| Mínimo | 0 | 19 | 3 | 4 | 4 |
| Desviación estándar | 2,18 | 75,79 | 49,95 | 7,88 | 7,07 |
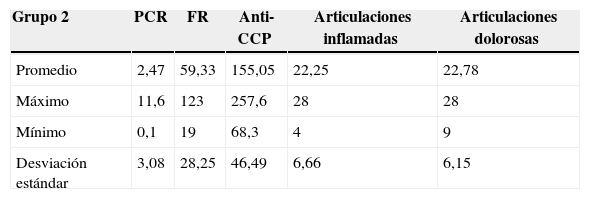
Análisis estadístico de los datos que conforman el grupo 2 de pacientes teniendo en cuenta la severidad de la enfermedad
| Grupo 2 | PCR | FR | Anti-CCP | Articulaciones inflamadas | Articulaciones dolorosas |
|---|---|---|---|---|---|
| Promedio | 2,47 | 59,33 | 155,05 | 22,25 | 22,78 |
| Máximo | 11,6 | 123 | 257,6 | 28 | 28 |
| Mínimo | 0,1 | 19 | 68,3 | 4 | 9 |
| Desviación estándar | 3,08 | 28,25 | 46,49 | 6,66 | 6,15 |
Al comparar los 2 grupos, se identificó que el promedio de articulaciones inflamadas y dolorosas entre estos fue similar, siendo en el grupo 1 de 23,26 y 24,08, respectivamente, y en el grupo 2 de 22,25 y 22,78, respectivamente, por lo cual estas 2 variables no son discriminantes. Por el contrario, el promedio de la variable anti-CCP del grupo 1 fue de 32,13 y del grupo 2 fue de 155,05, lo que puede indicar una enfermedad más activa en el grupo 2 con respecto al grupo 1. También, al analizar las desviaciones estándar del anti-CCP se puede inferir que esta variable puede llegar a ser discriminante.
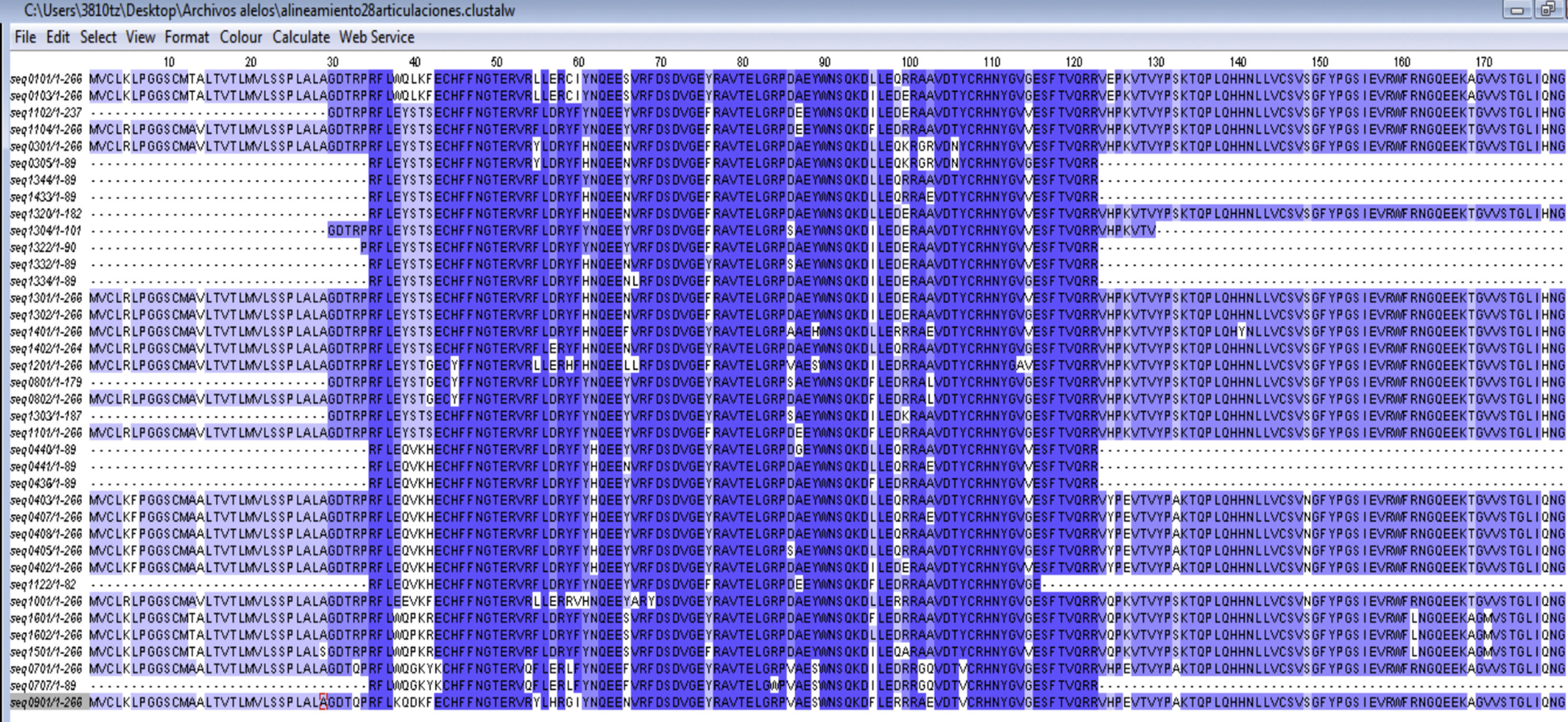
Por otro lado, para la identificación de endofenotipos se realizó el alineamiento de las secuencias de aminoácidos de los distintos alelos correspondientes a pacientes con un desenlace en común. Esto se hizo usando clustalw. Para este caso se seleccionaron los alelos de los pacientes con 28 articulaciones comprometidas como criterio de severidad y se realizó el alineamiento de las secuencias correspondientes. Como resultado se encontraron secuencias comunes, además de las secuencias de la teoría del EC.
Se realizó primero la identificación de la identidad entre las secuencias de aminoácidos que corresponden a pacientes con 28 articulaciones comprometidas; el resultado se muestra en la figura 9.

En la figura 10 se observa que las secuencias de aminoácidos QVKH y WQPKR son comunes en pacientes que tienen 28 articulaciones comprometidas, lo cual indica que están asociadas con este desenlace.

En el recuadro rojo se resalta el área correspondiente al EC. En este estudio se logró determinar que, además de las secuencias de aminoácidos comunes entre pacientes con AR, también existen otras secuencias de aminoácidos comunes entre pacientes, las cuales pueden ser candidatas para la posible identificación de desenlaces de la enfermedad.
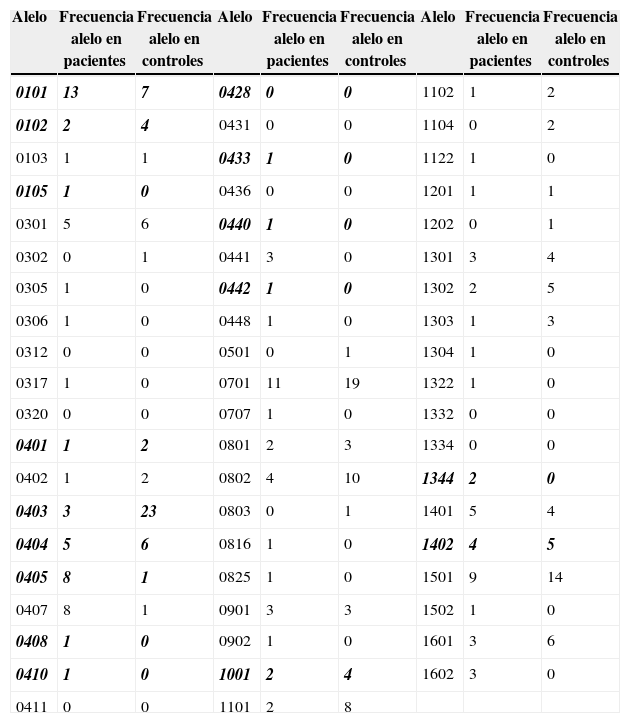
Se realizó un análisis de los datos tipificados en el HLA DRB1 y la información clínica de los pacientes con AR y de los controles. Esto se realizó a partir de la hipótesis del EC, para lo cual se calculó la frecuencia de cada alelo entre los pacientes y los controles. En la tabla 13 se muestran los resultados obtenidos. De las 138 personas que participaron en el estudio se identificaron 59 alelos diferentes para el HLA DRB1, de los cuales 8 son homocigotos y pertenecen a 4 pacientes (0101, 0103, 0441, 0701) y 4 controles (0701, 1101, 0802, 1303).
Resultados del análisis de alelos HLA DRB1 en pacientes y controles. Los alelos 0101, 0102, 0105, 0401, 0403, 0404, 0405, 0408, 0410, 0428, 0433, 0440, 0442, 1001, 1344, 1402 corresponden al epítope compartido
| Alelo | Frecuencia alelo en pacientes | Frecuencia alelo en controles | Alelo | Frecuencia alelo en pacientes | Frecuencia alelo en controles | Alelo | Frecuencia alelo en pacientes | Frecuencia alelo en controles |
|---|---|---|---|---|---|---|---|---|
| 0101 | 13 | 7 | 0428 | 0 | 0 | 1102 | 1 | 2 |
| 0102 | 2 | 4 | 0431 | 0 | 0 | 1104 | 0 | 2 |
| 0103 | 1 | 1 | 0433 | 1 | 0 | 1122 | 1 | 0 |
| 0105 | 1 | 0 | 0436 | 0 | 0 | 1201 | 1 | 1 |
| 0301 | 5 | 6 | 0440 | 1 | 0 | 1202 | 0 | 1 |
| 0302 | 0 | 1 | 0441 | 3 | 0 | 1301 | 3 | 4 |
| 0305 | 1 | 0 | 0442 | 1 | 0 | 1302 | 2 | 5 |
| 0306 | 1 | 0 | 0448 | 1 | 0 | 1303 | 1 | 3 |
| 0312 | 0 | 0 | 0501 | 0 | 1 | 1304 | 1 | 0 |
| 0317 | 1 | 0 | 0701 | 11 | 19 | 1322 | 1 | 0 |
| 0320 | 0 | 0 | 0707 | 1 | 0 | 1332 | 0 | 0 |
| 0401 | 1 | 2 | 0801 | 2 | 3 | 1334 | 0 | 0 |
| 0402 | 1 | 2 | 0802 | 4 | 10 | 1344 | 2 | 0 |
| 0403 | 3 | 23 | 0803 | 0 | 1 | 1401 | 5 | 4 |
| 0404 | 5 | 6 | 0816 | 1 | 0 | 1402 | 4 | 5 |
| 0405 | 8 | 1 | 0825 | 1 | 0 | 1501 | 9 | 14 |
| 0407 | 8 | 1 | 0901 | 3 | 3 | 1502 | 1 | 0 |
| 0408 | 1 | 0 | 0902 | 1 | 0 | 1601 | 3 | 6 |
| 0410 | 1 | 0 | 1001 | 2 | 4 | 1602 | 3 | 0 |
| 0411 | 0 | 0 | 1101 | 2 | 8 |
Los datos en cursiva y negrilla corresponden a alelos del epítope compartido con su respectiva frecuencia en pacientes y controles.
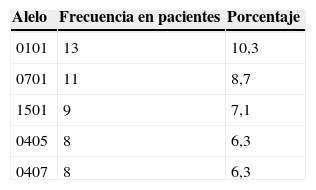
Los alelos más frecuentes en los pacientes se presentan en la tabla 14.
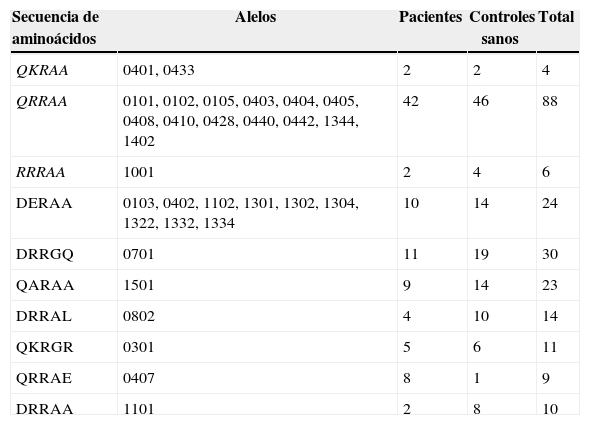
Las secuencias QKRAA, QRRAA y RRRAA, que corresponden a los aminoácidos de los alelos del EC, se repitieron 98 veces en los datos analizados. Esto significa que estas secuencias hacen parte de las reportadas en la literatura como EC.
En varios estudios se ha demostrado que algunas variantes genéticas se asocian con aumento del riesgo de desarrollar AR. Como en este estudio, en el cual un análisis de la interacción de polimorfismos se utilizó para analizar los polimorfismos de nucleótido único (SNP) que contribuyen a la AR, los pares de SNP adquiridos se utilizaron para construir una red SNP-SNP. Se realizó el análisis de cada módulo de genes que pueden ser utilizados para investigar la función de genes agrupados, para una mejor comprensión de la patogénesis de la AR15.
A partir de esta información, para cada uno de los alelos en estudio se encontraron las secuencias que se muestran en la tabla 15.
Correspondencia entre alelos y secuencias de aminoácidos. Las secuencias de aminoácidos en QKRAA, QRRAA y RRRA corresponden al epítope compartido
| Secuencia de aminoácidos | Alelos | Pacientes | Controles sanos | Total |
|---|---|---|---|---|
| QKRAA | 0401, 0433 | 2 | 2 | 4 |
| QRRAA | 0101, 0102, 0105, 0403, 0404, 0405, 0408, 0410, 0428, 0440, 0442, 1344, 1402 | 42 | 46 | 88 |
| RRRAA | 1001 | 2 | 4 | 6 |
| DERAA | 0103, 0402, 1102, 1301, 1302, 1304, 1322, 1332, 1334 | 10 | 14 | 24 |
| DRRGQ | 0701 | 11 | 19 | 30 |
| QARAA | 1501 | 9 | 14 | 23 |
| DRRAL | 0802 | 4 | 10 | 14 |
| QKRGR | 0301 | 5 | 6 | 11 |
| QRRAE | 0407 | 8 | 1 | 9 |
| DRRAA | 1101 | 2 | 8 | 10 |
Se realizó un alineamiento entre las distintas secuencias de aminoácidos de los alelos. El resultado de este alineamiento se muestra en la figura 11.

Con los datos utilizados en el presente estudio se confirma la hipótesis del EC en los pacientes colombianos con AR, esto a partir de que los alelos más frecuentes y que corresponden a las secuencias QKRAA, QRRAA y RRRAA están presentes en los pacientes con AR, se logra identificar que el alelo 0101, implicado en la AR, es el más frecuente en pacientes con AR.
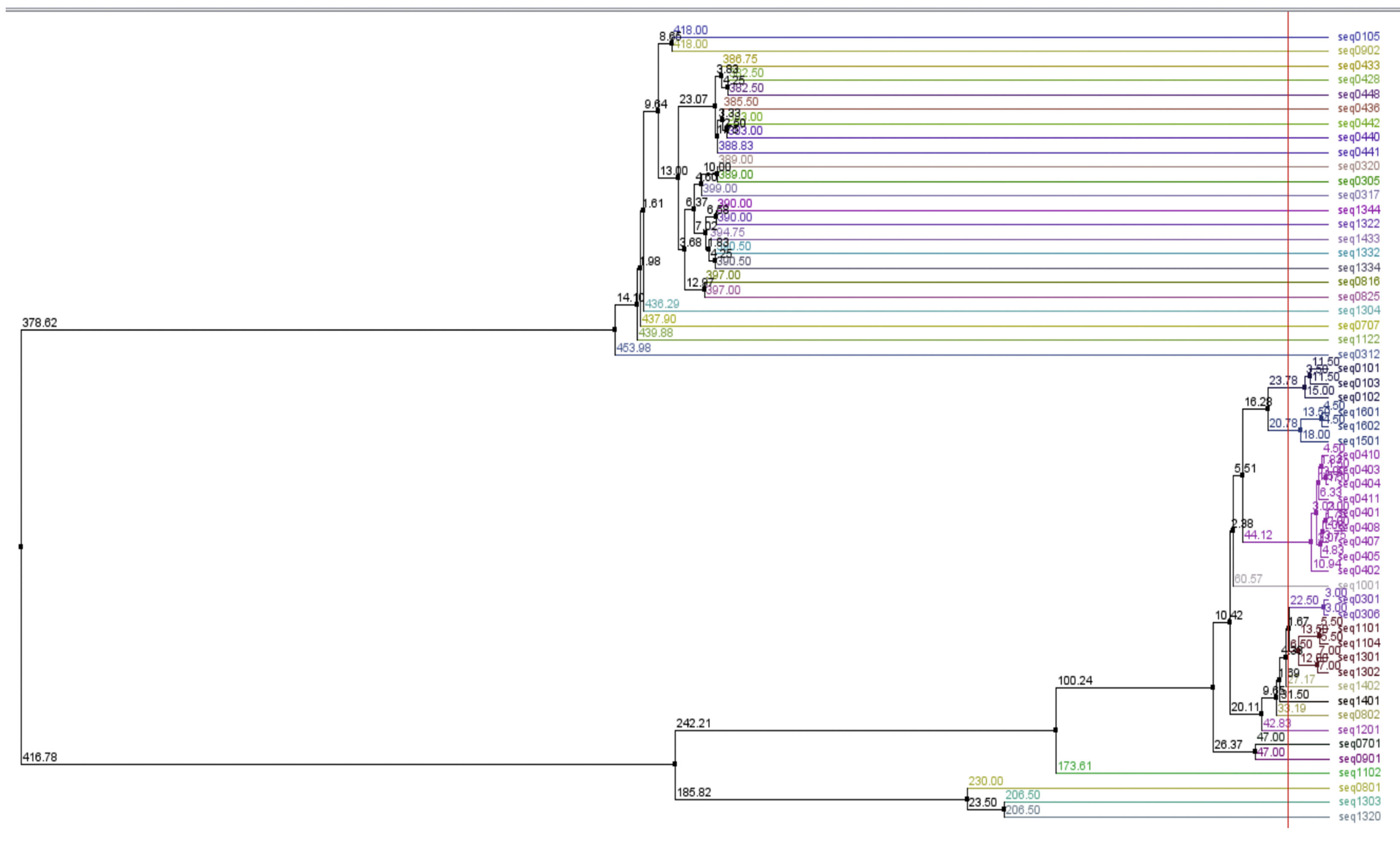
Finalmente, se creó un árbol entre los alelos de los pacientes con AR por el método de distancia promedio (average distance) usando Blosum62 con el fin de buscar cercanía genética entre los alelos, para posteriormente asociarlos con lo reportado en la literatura. En la figura 12 se presenta el árbol encontrado, el cual fue obtenido usando Jalview.

En la figura 12 se observan los grupos que se forman a partir de información de los pacientes. Allí se identifican algunos grupos que verifican lo que ha sido reportado en la literatura sobre criterios de severidad. Particularmente se forman grupos con riesgo alto que corresponden a los alelos 0401 y 1303; riesgo intermedio 0101, 0102, 0404, 0405, 0408, 1001, 1402; y riesgo bajo 0103, 0402, 03, 0403, 0407, 0411, 07, 08, 09, 1101, 1102, 1103, 1104, 12, 1301, 1302, 1323, 1401, 1404, 15, 16.
La clasificación propuesta en un estudio previo fue la siguiente, el HLA-DRB1 se dividió primero en 2 grupos de acuerdo con la presencia o ausencia de la secuencia RAA en la posición 72-74, que se denota como S y alelos X, respectivamente. El grupo S incluye alelos que se clasifican como EX en la primera clasificación. Los alelos S se puede dividir posteriormente en 4 grupos de acuerdo con el aminoácido en la posición 71: una alanina (A), un ácido glutámico (E), una lisina (K), o una arginina (R). Cuatro diferentes alelos fueron definidos en la nueva clasificación, así: S1 para ARAA y ERAA, S2 para KRAA, S3 para RRAA y X para todos los motivos no RAA. Por otra parte, el estudio indica que el aminoácido en la posición 70 puede influir en la susceptibilidad a la AR; de hecho, un ácido aspártico (D) se supone que es de protección en comparación con la presencia de una glutamina (Q) o una arginina (R). Por lo tanto, 2 alelos adicionales se definieron: S3D para DRRAA y S3P para QRRAA o RRRAA. Alelos S2 no podrían ser divididos debido a su pequeña frecuencia16,17.
En la tabla 16 se presentan los grupos obtenidos por cercanía genética a partir de la secuencia de aminoácidos correspondiente a cada alelo, los cuales son comparables con resultados previos obtenidos en otros estudios.
DiscusiónMediante técnicas computacionales, como las redes neuronales, utilizando 5 variables, se logró clasificar a pacientes con AR y controles sanos; utilizando los criterios del Colegio Americano de Reumatología se logró mejorar el desempeño de los modelos. En el caso de la red neuronal se consiguió clasificar adecuadamente el 89,28% de los datos. Con la red bayesiana con 5 variables se clasificó adecuadamente el 92,85% de los datos. Por otro lado, la red neuronal utilizando 11 variables clasificó adecuadamente el 89,28% y la red bayesiana clasificó adecuadamente el 92,85% de los datos.
Los resultados obtenidos con k-means con el grupo de 138 individuos permitieron determinar que el grupo 1 presentaba una susceptibilidad mayor a presentar AR dada por los resultados de las variables analizadas.
Posteriormente, se realizó un nuevo agrupamiento de pacientes para determinar la severidad de la enfermedad. Al utilizar el coeficiente silueta se determinó que el mejor agrupamiento se logra con 2 grupos. Al comparar los 2 grupos, se identificó que el promedio de articulaciones inflamadas y dolorosas fue similar, por lo cual, estas 2 variables no son discriminantes. En contraste, la variable anti-CCP resultó discriminante para la condición de severidad de la enfermedad. Por otro lado, los valores para la variable FR son cualitativamente positivos, lo cual solo indica que los individuos pertenecientes a estos grupos presentan una actividad de la enfermedad. Algo similar se puede observar para las articulaciones inflamadas y dolorosas.
Adicionalmente, en la red bayesiana se identificaron relaciones significativas entre las variables, tales como la relación entre el número de articulaciones inflamadas y dolorosas con las variables serológicas y las variables genéticas. Este hallazgo está acorde con los resultados obtenidos en el estudio de Li et al.18, donde analizaron las relaciones existentes entre los fenotipos y biomarcadores en un estudio de AR. Por lo cual, en este estudio a través de los métodos computacionales mencionados se encontraron relaciones de dependencia entre estas variables, principalmente usando los modelos bayesianos. Las tablas de los valores de las probabilidades correspondientes indican la posibilidad de que una persona presente AR.
Según algunos estudios, las redes bayesianas proporcionan un marco conveniente para la sistematización de datos para deducir un orden de probabilidad y modelos de variables que interactúan en enfermedades como la AR. Estos estudios se han concentrado en la estimación de las relaciones estructurales en el sistema en estudio o en la identificación de las asociaciones de la enfermedad con los polimorfismos genéticos. Por esto las redes bayesianas se pueden utilizar para predecir el resultado de las intervenciones específicas en esta enfermedad19.
ConclusionesEn este trabajo se realizó el análisis de un conjunto de datos de pacientes con AR aplicando métodos de inteligencia computacional, con el fin de tratar de identificar endofenotipos para la enfermedad. Los datos incluían información genética, clínica y serológica. También se trataron de obtener marcadores para determinar la severidad de la enfermedad en los pacientes.
Se identificaron las relaciones de dependencia entre algunas de las variables a través de una red bayesiana. Específicamente se estableció que las variables anti-CCP y el FR están implicadas de manera significativa en la enfermedad. También, se identificaron endofenotipos relacionados con la severidad de la enfermedad en pacientes; particularmente se identificaron secuencias de aminoácidos comunes entre pacientes con el mismo desenlace.
Se concluyó que utilizando técnicas de inteligencia computacional y bioinformática fue posible desarrollar modelos computacionales que, usando información genética y clínica, logran clasificar entre pacientes y controles sanos, así como también clasificar según criterios de severidad a los pacientes.
Trabajo futuroSe propone trabajar en la búsqueda de más secuencias comunes entre los distintos pacientes con grados de severidad similares y para ello se podrán utilizar otras variables como son niveles elevados del FR, niveles elevados de anticuerpos anti-CCP, HAQ elevados y otras posibles variables que se pueden cuantificar.
Se recomienda en un trabajo futuro desarrollar una aplicación web que permita automatizar la metodología de análisis aplicada en esta investigación, así como realizar trabajos futuros con un número mayor de pacientes.
Finalmente, se recomienda implementar y validar otras técnicas de inteligencia computacional, tales como máquinas de soporte vectorial o técnicas como boosting y bagging, para clasificar y diferenciar entre pacientes y controles sanos.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que los procedimientos seguidos se conformaron a las normas éticas del comité de experimentación humana responsable y de acuerdo con la Asociación Médica Mundial y la Declaración de Helsinki.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
