


La cromatografía líquida de alta resolución o high-performance liquid chromatography (HPLC) es un principio de medida que permite separar los componentes de una muestra (analitos) en función de su distribución entre dos fases inmiscibles entre sí: una fase estacionaria (un sólido o un líquido adsorbido sobre un soporte sólido) y una fase móvil (un líquido). En este tipo de cromatografía, para que tenga lugar el proceso físico-químico de separación, la muestra es inyectada en el seno de la fase móvil, en la que es soluble, e impulsada a una elevada presión a través de una columna que contiene la fase estacionaria. Una vez separados los diferentes analitos en el sistema cromatográfico (eluatos), y en función de sus propiedades fisico-químicas, estos pueden ser identificados o cuantificados empleando alguno de los múltiples sistemas de detección existentes.
Debido a la elevada capacidad para separar, aislar, identificar y cuantificar que presenta la HPLC, los procedimientos de medida basados en esta tecnología (de ahora en adelante, procedimientos cromatográficos) llevan años aplicándose en los centros de investigación, en la industria farmacéutica, en las empresas del diagnóstico in vitro o en los laboratorios clínicos para el desarrollo de nuevos fármacos o para la identificación o la medición de nuevas propiedades biológicas1,2. En los últimos años, la aplicación de la HPLC en los laboratorios clínicos ha adquirido un papel más relevante debido, principalmente, a la aparición de sistemas de medida que combinan la HPLC con sistemas de detección específicos y con una elevada capacidad de detección como son los espectrómetros de masas3–6. Pese a ello, en la actualidad, son pocos los laboratorios clínicos españoles que emplean procedimientos cromatográficos debido, principalmente, a la falta de total automatización de los mismos, a la necesidad de personal que previamente haya sido específicamente habilitado para su manejo y a su relativo elevado coste. Adicionalmente, existen un número reducido de empresas que desarrollan procedimientos cromatográficos para el diagnóstico in vitro y con la marca «CE», lo que obliga en muchos casos a los laboratorios clínicos a realizar el desarrollo y la validación de los mismos7.
Objeto y campo de aplicaciónEl objeto de este documento es describir el proceso que deben realizar los laboratorios clínicos para el desarrollo de los procedimientos cromatográficos.
El campo de aplicación de este documento abarca los procedimientos cromatográficos que permiten la medición de propiedades biológicas correspondientes a escalas racionales (propiedades «cuantitativas») y que utilicen como principio de medida la HPLC acoplada a cualquier sistema de detección.
En este documento son aplicables los términos y definiciones relacionados con la cromatografía recogidos en las diferentes guías internacionales de la Federación Internacional de Química Pura y Aplicada (IUPAC)8–11, el Instituto para la Normalización de Laboratorios Clínicos (CLSI)12,13, la Agencia Europea del Medicamento (EMA)14 y la Administración de Alimentos y Medicamentos de los Estados Unidos (FDA)15.
Desarrollo de procedimientos de medida cromatográficosExisten diferentes aspectos que deben considerarse en el desarrollo de un procedimiento cromatográfico que se inicia con la recogida de información sobre el analito a estudiar hasta el proceso final de validación del mismo16–25. En este apartado se describen las principales etapas necesarias para el desarrollo de un procedimiento cromatográfico.
Descripción y recopilación de información relacionada con el analitoEl primer aspecto a considerar es recoger toda la información relacionada con el analito a estudiar, como la estructura química, la masa molar, la polaridad, la solubilidad y la constante de disociación ácida (pKa), entre otros. Esta información es importante para la selección de las condiciones cromatográficas y el tipo de sistema de detección a emplear.
El conocimiento de la masa molar del analito es imprescindible cuando se trabaja con la HPLC acoplada a la espectrometría de masas (HPLC-MS) o a la espectrometría de masas en tándem (HPLC-MS/MS), ya que se basan principalmente en la obtención de iones a partir de moléculas en fase gaseosa que son posteriormente separados según su relación masa/carga.
Existen diversos recursos en Internet (webs) que permiten conocer las principales propiedades fisico-químicas de un analito. Son un ejemplo:
- -
La base de datos PubChem del Instituto Nacional de la Salud de los Estados Unidos: https://pubchem.ncbi.nlm.nih.gov/
- -
La base de datos ChemSpider de la Real Sociedad de Química: http://www.chemspider.com/
- -
Las bases de datos ChEMBL y ChEBI del Instituto Europeo de Bioinformática: https://www.ebi.ac.uk/chembl/
- -
- -
La base de datos ZINC de la Universidad de California, San Francisco, Estados Unidos: http://zinc.docking.org/
- -
La base de datos de fármacos Drug bank: http://www.drugbank.ca/
- -
La base de datos de fármacos ACToR de la Agencia de Protección Ambiental de Estados Unidos: http://actor.epa.gov/actor/faces/ACToRHome.jsp
Antes de iniciar el desarrollo de un procedimiento cromatográfico, el laboratorio clínico debe decidir el objetivo de las mediciones. Este aspecto es fundamental para escoger el tipo de separación cromatográfica a utilizar, así como para conocer si el procedimiento a desarrollar será más o menos complejo y costoso. Los principales aspectos a considerar son16–21:
- -
Decidir si el procedimiento va a ser empleado para la caracterización o cuantificación de uno o varios analitos de una muestra desconocida.
- -
Establecer si el objetivo principal va a ser la medición de la concentración del analito o solo conocer la presencia del mismo en una muestra determinada («determinación cualitativa»).
- -
Determinar el número de analitos a estudiar.
- -
Decidir si será necesario separar todos los componentes de la muestra.
- -
Conocer con qué tipo de especímenes se va a trabajar (p. ej.: plasma, orina, suero, etc.).
- -
Conocer la carga de trabajo aproximada de las muestras que van a ser procesadas.
- -
Establecer la periodicidad del procesamiento de las muestras (p. ej.: diaria, quincenal, mensual, etc.).
- -
Tener en cuenta el tipo de cromatógrafo líquido de alta eficacia y el sistema de detección con el que cuenta el laboratorio.
El tratamiento y la preparación de las muestras biológicas es un proceso esencial. Su importancia radica en la necesidad de concentrarla o eliminar los distintos componentes de la muestra que pueden interferir y obtener así, una apropiada separación y eficacia cromatográficas. La presencia de proteínas, lípidos, sales y componentes celulares pueden interferir en la medición de la magnitud en estudio (obtención de señales erróneas en el detector y picos cromatográficos asimétricos, resolución inapropiada de picos cromatográficos, falta de reproducibilidad de los tiempos de retención, una menor capacidad de detección, etc.), así como condicionar el buen funcionamiento del sistema cromatográfico (obstrucción del inyector, fritas (p. ej. ubicadas en la entrada de la columna), detector, cánulas, tubos, válvulas, etc.)13,14,17–21.
Los principales procedimientos de preparación de la muestra son la filtración, la centrifugación, la dilución e inyección, la precipitación de proteínas, la extracción líquido-líquido y la extracción en fase sólida. La elección de un procedimiento u otro dependerá del tipo de muestra biológica con la que se trabaje y de las propiedades físico-químicas del analito, siendo incluso necesario, algunas veces, la combinación de dos o más de estos procedimientos.
Pese a las ventajas que supone la aplicación de estos procedimientos, la mayoría de ellos pueden dar lugar a una pérdida parcial del analito y, por otro lado, no todos permiten eliminar totalmente los diferentes interferentes de la muestra.
Dilución e inyecciónEs el procedimiento más simple y se suele emplear para las muestras biológicas relativamente poco complejas (orina, líquido cefalorraquídeo). Consiste en diluir la muestra con un disolvente adecuado e inyectar la muestra diluida en el sistema cromatográfico. Este procedimiento permite reducir el posible efecto negativo de los distintos interferentes de la muestra sobre la medición de la magnitud en estudio pero, al no eliminarlos, pueden acumularse en el sistema cromatográfico y provocar un mal funcionamiento del mismo a lo largo del tiempo.
Precipitación de proteínasEs el procedimiento más empleado debido a su simplicidad y al reducido número de reactivos que requiere. Consiste en adicionar un reactivo químico particular que reduce notablemente la solubilidad de las proteínas en la muestra haciendo que precipiten. Posteriormente, este precipitado es eliminado por filtración o centrifugación dejando un sobrenadante que es inyectado directamente al sistema cromatográfico. Adicionalmente, si se requiere, el sobrenadante puede ser diluido con un tampón o un disolvente apropiado (habitualmente la fase móvil) o evaporado (p. ej. empleando nitrógeno) hasta la sequedad y seguidamente diluido con un tampón o un disolvente apropiado.
Dependiendo del reactivo de precipitación utilizado, de las condiciones experimentales y de las propiedades fisico-químicas de los componentes de la muestra, se pueden llegar a eliminar hasta el 98% de las proteínas de la muestra.
El procedimiento de precipitación de proteínas más ampliamente utilizado se basa en la adición de un disolvente orgánico (principalmente acetonitrilo o metanol) a la muestra en una proporción determinada —4:1, 3:1 (la más empleada), 2:1—. También se usan otros reactivos químicos (p. ej.: ácido tricloroacético, hidróxido de zinc, sulfato de zinc o cloruro de magnesio), individualmente o conjuntamente con disolventes orgánicos.
Hay que tener presente que en este procedimiento también se diluye el analito y, además, no se eliminan todos los componentes de la muestra (únicamente se eliminan la mayoría de las proteínas y los componentes celulares), quedando en el sobrenadante otros componentes —sales y lípidos— que pueden interferir en la medición de la magnitud biológica o en el buen funcionamiento del sistema cromatográfico. En determinados casos, puede ser necesario realizar otros procedimientos de extracción que permitan reducir estos componentes biológicos.
Extracción líquido-líquidoLa extracción líquido-líquido es un procedimiento de preparación de la muestra que se fundamenta en la transferencia del analito entre dos líquidos inmiscibles (fase acuosa y fase orgánica), la cual depende de su solubilidad entre ambos líquidos. De modo general, a la muestra que contiene el analito (fase acuosa) se adiciona un líquido orgánico inmiscible (fase orgánica) que actúa como extractante. Después de un periodo de agitación, el analito se distribuye entre las dos fases hasta alcanzar una situación de equilibrio. Seguidamente, se deja reposar el sistema el tiempo necesario para que se produzca la separación de las fases o bien se centrifuga. La transferencia del analito se rige por la ley de distribución de Nernst que establece que, a presión y temperatura constantes, la relación de concentración de un analito entre dos fases inmiscibles es constante. Esta ley se materializa con la constante de distribución (KD):

donde [A]o representa la concentración de analito en la fase orgánica y [A]a la concentración de analito en la fase acuosa. Cada analito tiene un valor característico de KD para un par de disolventes concretos. Cuanto mayor es la KD, mayor es la eficacia de la extracción, de modo que a la hora de desarrollar un procedimiento líquido-líquido se debe tener una idea clara de la polaridad del analito y de los disolventes empleados. Si un analito es poco soluble en agua, su extracción se verá más favorecida por el empleo de disolventes apolares que empleando disolventes polares. Los disolventes orgánicos que suelen utilizarse son hexano, diclorometano, 1,2-dicloroetano, metil-tert-butil éter, entre otros.
En este procedimiento también se diluye el analito, aunque puede concentrarse mediante evaporación del disolvente orgánico y reconstitución posterior con un disolvente apropiado. Además, no se eliminan todos los componentes de la muestra (únicamente se eliminan componentes celulares, las sales y algunas proteínas) quedando en el sobrenadante otros componentes como los lípidos, que pueden interferir en la medición de la magnitud biológica.
Extracción en fase sólidaLa extracción en fase sólida o extracción sólido-líquido es un procedimiento en el que el analito se separa en función de la afinidad relativa entre la matriz de la muestra y un sólido que contiene una fase sólida (sorbente). La interacción sorbente-analito puede responder a diferentes mecanismos físico-químicos, entre los que cabe destacar la adsorción, partición, intercambio iónico, exclusión molecular, afinidad o una combinación de ellos. Esta situación permite que la extracción en fase sólida sea muy versátil y selectiva.
Generalmente, comprende cinco etapas:
- -
Activación o acondicionamiento de la fase sólida. Consiste en la solvatación del sólido empleando un disolvente orgánico.
- -
Equilibrado de la fase sólida. En esta etapa se emplea un disolvente similar a la matriz de la muestra que permite eliminar el exceso de disolvente acondicionador.
- -
Adición de la muestra para conseguir la retención del analito por la fase sólida. En este paso aquellos potenciales interferentes que no presentan afinidad alguna por la fase sólida son descartados y eliminados.
- -
Lavado de la fase sólida. En esta etapa se usa un disolvente o mezcla de disolventes para la eliminación selectiva de los interferentes que hayan podido quedar retenidos débilmente a la fase sólida.
- -
Elución del analito. Mediante la adición de un disolvente adecuado se consigue debilitar la interacción y unión del analito a la fase sólida permitiendo la recuperación selectiva del mismo.
En este procedimiento no se produce una dilución de la muestra (o lo hace en una proporción baja) y permite eliminar prácticamente la totalidad de los interferentes, proporcionando una mayor reproducibilidad que el resto de los procedimientos de extracción descritos.
Selección del tipo de cromatografía líquida y la modalidad de trabajoPara llevar a cabo una adecuada separación y eficacia cromatográficas, se requiere que exista un equilibrio entre las fuerzas intermoleculares de los tres participantes activos en el proceso de separación: el analito, la fase móvil y la fase estacionaria. Por consiguiente, cuando se desarrolla un procedimiento cromatográfico, se debe seleccionar el tipo de cromatografía líquida (de adsorción, de reparto, de intercambio iónico, de exclusión molecular o de afinidad) atendiendo a cuáles son las interacciones que existen entre el analito y las diferentes fases22,23.
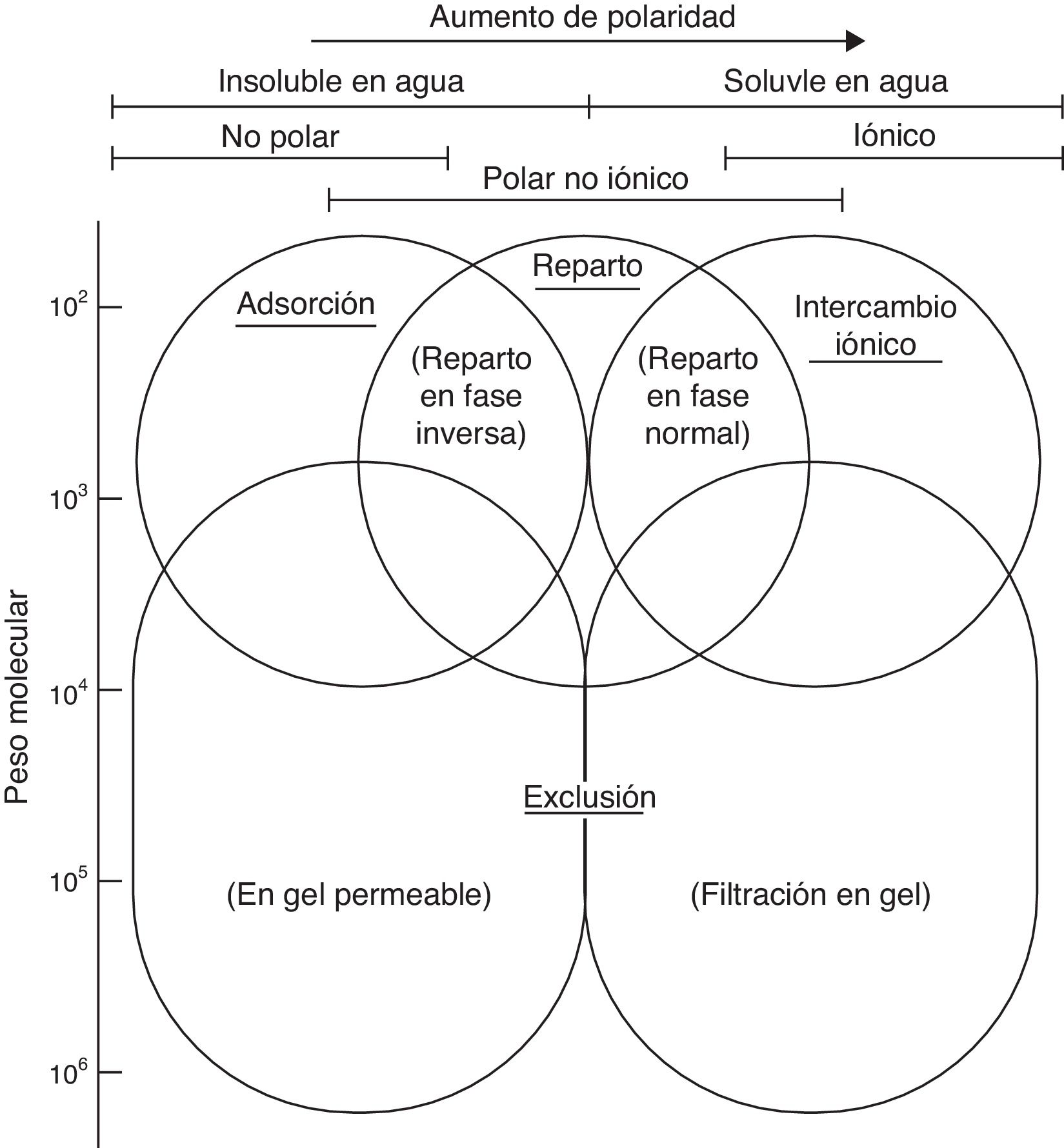
En la figura 1 se muestra un esquema relacionado con la selección del tipo de cromatografía líquida en función de las propiedades físico-químicas del analito.

Tipos de cromatografía líquida a emplear en función de la masa molar y la polaridad del analito. Adaptada de Skoog22.
En la cromatografía de reparto, existen dos modalidades cromatográficas de trabajo, la cromatografía en fase normal y la cromatografía en fase inversa. La cromatografía en fase normal se caracteriza por utilizar una fase móvil apolar o poco polar y una fase estacionaria polar. En esta modalidad, los analitos más polares quedan más retenidos en la fase estacionaria mientras que los menos polares eluyen primero. Por el contrario, en la cromatografía en fase inversa se emplean fases móviles polares y fases estacionarias apolares o poco polares. En este caso, los analitos más polares eluyen en primer lugar mientras que los menos polares quedan más retenidos.
La cromatografía de reparto en la modalidad de fase inversa, al ser más versátil que otros tipos de cromatografías debido a su mecanismo de separación, actualmente es la más utilizada en las ciencias de laboratorio clínico y la que debería emplearse cuando se pretende desarrollar un procedimiento cromatográfico. Sin embargo, para la separación de ciertos analitos —que presentan unas propiedades físico-químicas muy específicas y diferenciadas— es aconsejable emplear otros tipos de cromatografía:
- •
Para analitos con elevada masa molar (p. ej., macromoléculas como enzimas y proteínas), es aconsejable utilizar la cromatografía de exclusión molecular, aunque esta dará lugar a una menor resolución cromatográfica.
- •
Para analitos que son iones inorgánicos (p. ej., hierro, zinc, nitritos, nitratos) o analitos fácilmente ionizables (p. ej., aminas biógenas, hemoglobinas), es preferible emplear la cromatografía de intercambio iónico.
- •
Para analitos que presentan varios estereoisómeros y presentan una polaridad elevada, es preferible emplear la cromatografía de adsorción.
- •
Para analitos que no pueden ser separados por otros tipos de cromatografía o bien son susceptibles de ser separados mediante reacciones biológicas específicas del tipo antígeno-anticuerpo (p. ej., inmunoglobulinas), enzima-sustrato (p. ej., enzimas), entre otras, es preferible emplear la cromatografía de afinidad.
La elección del sistema cromatográfico dependerá, entre otros factores, de la naturaleza química del analito a estudiar (es decir, de sus propiedades físico-químicas), de las posibles interferencias que puedan existir después del proceso de preparación de la muestra, de la capacidad de detección (detectabilidad) que se requiera y de los detectores cromatográficos disponibles en el laboratorio clínico16–24.
Siempre que sea posible, el laboratorio clínico debería seleccionar detectores cromatográficos con elevada versatilidad, tales como los de absorción molecular, los de índice de refracción y los espectrómetros de masas. Generalmente, los detectores cromatográficos de absorción molecular suelen ser los más empleados dada la naturaleza orgánica de los analitos que se estudian en los laboratorios16–24. Además, suelen tener un coste asumible. En el caso de que los analitos no absorban la radiación electromagnética o presenten una baja absorción (p. ej.: glúcidos, ácidos grasos y algunos lípidos), es recomendable utilizar los detectores de índice de refracción. Este tipo de detectores presentan una menor sensibilidad metrológica, capacidad de detección y selectividad que los de absorción, y su coste suele ser similar. Además, estos no permiten trabajar en la modalidad en gradiente.
De todos estos detectores, el más versátil y selectivo es el espectrómetro de masas ya que, al basarse en la medición de propiedades de la materia como la masa y la carga, permite prácticamente la detección de cualquier tipo de analito. Su principal desventaja es su elevado coste16–24.
Además de los detectores mencionados, existen otros que son más selectivos, sensibles y con una mayor capacidad de detección (aunque menos que los espectrómetros de masas), como son los detectores de fluorescencia y los electroquímicos. Los detectores de fluorescencia son los que presentan mayor sensibilidad y capacidad de detección16–24. No obstante, para poder utilizar un detector de fluorescencia es necesario que los analitos presenten la capacidad de emitir fluorescencia o bien, que la emitan cuando se les hace reaccionar con otras sustancias químicas. Los detectores electroquímicos es recomendable emplearlos cuando se estudian analitos con propiedades oxidantes o reductoras, como por ejemplo las aminas biógenas (catecolaminas, metanefrinas, vanilmandelato, 5-hidroxiindolilacetato, entre otras). Este tipo de detectores han de ser utilizados con precaución ya que presentan una elevada sensibilidad y suelen ser susceptibles a interferencias eléctricas producidas por la propia fuente de alimentación eléctrica. El coste de los detectores fluorimétricos y electroquímicos suele ser similar o ligeramente superior a los de absorción o de índice de refracción16–24.
Optimización de la separación cromatográficaDurante el proceso de optimización de la separación cromatográfica, además de la necesidad de conocer las propiedades físico-químicas del analito, deben considerarse diferentes aspectos relacionados con la fase móvil y la fase estacionaria16–21 que se describen a continuación.
Composición de la fase móvilEl conocimiento de la polaridad del analito permite seleccionar los disolventes a emplear en la fase móvil. Así, la selección de la fase móvil debe llevarse a cabo en base a su solubilidad con el analito. Además, el analito no debe reaccionar con ninguna de las sustancias que componen el disolvente.
Existe una estrecha relación entre el tiempo de retención o elución de los analitos con la composición de la fase móvil como consecuencia del efecto de la polaridad. De hecho, en la HPLC de reparto de fase inversa, el tiempo de elución de los analitos puede ser alterado modificando la polaridad de la fase móvil, es decir, variando el tipo de disolventes a utilizar y el porcentaje empleado de cada uno de ellos. Esta es la manera más sencilla para intentar mejorar la resolución cromatográfica de dos componentes de una muestra cuyos picos cromatográficos se solapan, o para disminuir el tiempo global de separación de los componentes cuando estos presentan tiempos de retención muy diferentes.
Un adecuado punto de partida para optimizar la separación cromatográfica sería emplear una mezcla de agua y un disolvente orgánico polar (metanol o acetonitrilo), con una mayor proporción de agua (p. ej.: cercano al 100%), y observar cómo varían los tiempos de elución y la resolución de los analitos al disminuir la proporción acuosa y aumentar la proporción del disolvente orgánico.
pH de la fase móvilEl ajuste del pH de la fase móvil y el conocimiento del pKa del analito tienen un papel muy importante para establecer las condiciones cromatográficas iniciales y su posterior optimización. La selección de un pH adecuado dará lugar a que el analito esté o no ionizado y, a menudo, esta situación conducirá a la obtención de picos simétricos, estrechos y puntiagudos en el proceso cromatográfico. La obtención de este tipo de picos es necesaria cuando la cromatografía es utilizada con una finalidad cuantitativa, ya que permitirá obtener mejores propiedades metrológicas (límites de detección y cuantificación más bajos, menores valores de imprecisión y sesgo), así como tiempos de retención cromatográficos reproducibles.
Las soluciones tampón deben añadirse a la fase móvil cuando el analito es un compuesto con propiedades iónicas. En la HPLC de reparto de fase inversa, los analitos de una muestra se separan en función de su polaridad de forma que los menos polares son más retenidos por la columna y viceversa. Cuando se ioniza un analito, aumenta su polaridad y, en consecuencia, presentará una menor retención en la columna. Los analitos de naturaleza ácida se ionizan cuando el pH es superior a su pKa y los analitos de naturaleza básica, cuando el pH es inferior a su pKa. Así, se obtendrá una mayor retención en la columna para los analitos ácidos cuando se trabaje a pH ácido y por debajo de su pKa; y una mayor retención también a pH básico y por encima de su pKa para los analitos básicos. La relación entre el analito ionizado y no ionizado —a un pH determinado— puede conocerse mediante la ecuación de Henderson-Hasselbach.
Teniendo en cuenta todas estas consideraciones, para obtener una mayor retención del analito y, en consecuencia, unos picos cromatográficos aceptables y una reproducibilidad adecuada, debe ajustarse el pH de la fase móvil dos unidades por debajo del pKa del analito, si este es de naturaleza ácida, y dos unidades por encima del pKa del analito si es de naturaleza básica. En estas condiciones, el 99% del analito no estará ionizado.
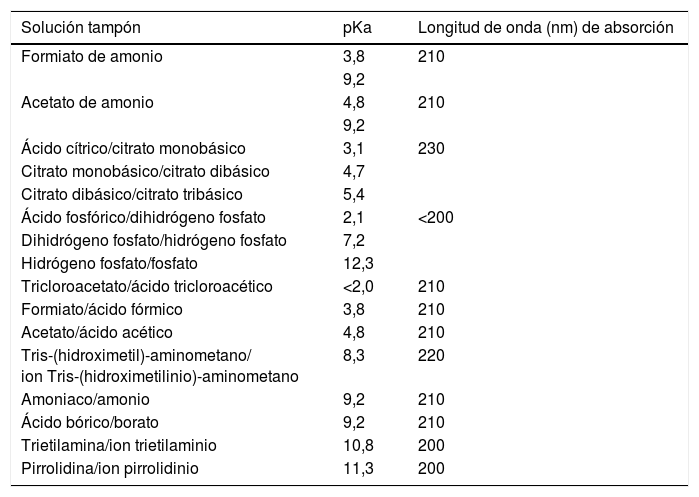
Las principales soluciones tampón y sus correspondientes pKa (a 25°C) se muestran en la tabla 1 El efecto óptimo de amortiguación de cada tampón se obtiene para el intervalo de pH comprendido entre pKa±1.
Principales soluciones tampón y sus correspondiente pKa y límites de absorción UV (ennm)
| Solución tampón | pKa | Longitud de onda (nm) de absorción |
|---|---|---|
| Formiato de amonio | 3,8 | 210 |
| 9,2 | ||
| Acetato de amonio | 4,8 | 210 |
| 9,2 | ||
| Ácido cítrico/citrato monobásico | 3,1 | 230 |
| Citrato monobásico/citrato dibásico | 4,7 | |
| Citrato dibásico/citrato tribásico | 5,4 | |
| Ácido fosfórico/dihidrógeno fosfato | 2,1 | <200 |
| Dihidrógeno fosfato/hidrógeno fosfato | 7,2 | |
| Hidrógeno fosfato/fosfato | 12,3 | |
| Tricloroacetato/ácido tricloroacético | <2,0 | 210 |
| Formiato/ácido fórmico | 3,8 | 210 |
| Acetato/ácido acético | 4,8 | 210 |
| Tris-(hidroximetil)-aminometano/ ion Tris-(hidroximetilinio)-aminometano | 8,3 | 220 |
| Amoniaco/amonio | 9,2 | 210 |
| Ácido bórico/borato | 9,2 | 210 |
| Trietilamina/ion trietilaminio | 10,8 | 200 |
| Pirrolidina/ion pirrolidinio | 11,3 | 200 |
Un aspecto a tener en cuenta durante la selección de una solución tampón es el sistema de detección con el que se va a trabajar. Si se emplea un sistema de absorción molecular en la zona del espectro visible, cualquiera de las soluciones tampón mencionadas puede ser utilizada, pero si se trabaja en la zona del ultravioleta, las soluciones tampón de citrato no son recomendables a no ser que se trabaje a longitudes de onda por encima de 230nm. De todos los tampones citados, los tampones fosfato son los más ampliamente empleados en la HPLC acoplada a la espectrometría de absorción molecular, dadas sus propiedades absortivas y amortiguadoras. Por otra parte, si se emplean sistemas de detección basados en la espectrometría de masas, deben utilizarse tampones volátiles (p. ej.: acetato de amonio, formiato de amonio y trietilamina) y evitar el uso de tampones no volátiles (p. ej.: fosfato y citrato).
Otra consideración importante es la concentración de tampón a utilizar. La solución tampón debe presentar una concentración suficientemente elevada como para poder controlar el pH de la fase móvil, pero no excesiva para evitar la posible precipitación de las sales del tampón en presencia de disolventes orgánicos. Habitualmente, una concentración apropiada de la solución tampón es aquella que está comprendida entre 10mmol/l y 50mmol/l. Los tampones fosfato, a concentraciones elevadas, deben evitarse ya que pueden precipitar. Este hecho se acentúa si se emplea una proporción elevada de disolvente orgánico en la fase móvil (> 50%).
Modalidad de elución de la fase móvilPor norma general, una elución isocrática —que mantiene constante la composición de la fase móvil durante todo el proceso cromatográfico— da lugar a separaciones cromatográficas más reproducibles que una elución en gradiente —que varía la composición de la fase móvil durante el proceso cromatográfico—. A pesar de ello, trabajar en la modalidad de elución isocrática presenta una serie de desventajas como son su bajo «poder de separación» —el número de analitos que pueden ser separados en un mismo proceso cromatográfico— y la obtención de picos cromatográficos más anchos en aquellos analitos que son más retenidos en la columna, afectando a la resolución cromatográfica. La elución isocrática se emplea para separaciones simples —de uno o pocos componentes— reservando la elución en gradiente para separaciones de mezclas más complejas, ya que permite aumentar significativamente el «poder de separación» como consecuencia de un aumento de la eficiencia cromatográfica (disminución de la anchura del pico), además de permitir disminuir el tiempo de la cromatografía y reducir el consumo de los disolventes que conforman la fase móvil.
Un adecuado punto de partida para optimizar la separación cromatográfica sería emplear la modalidad de elución isocrática, a no ser que se requiera separar un número elevado de analitos o bien reducir significativamente el tiempo de la cromatografía, en cuyo caso se debería trabajar en la modalidad de elución en gradiente.
Influencia de la temperatura de la fase móvilLa mayoría de los sistemas cromatográficos incluyen un compartimento específico para columnas que permite seleccionar la temperatura de trabajo de la fase móvil. La temperatura influye considerablemente en diferentes parámetros cromatográficos como la solubilidad, la difusividad, la viscosidad y la polaridad de la fase móvil.
A elevadas temperaturas (>40°C), la viscosidad de la fase móvil se reduce y la velocidad de difusión aumenta, provocando que la tasa de transferencia de masa del analito entre la fase estacionaria y la fase móvil se incremente y, consecuentemente, la eficacia, la resolución y la separación cromatográficas. Así, se puede trabajar con flujos de fase móvil ligeramente mayores que con los que se trabajaría a temperatura ambiente, hecho que permite acortar sustancialmente el tiempo de análisis sin perder eficacia.
Un punto de partida adecuado para optimizar la separación cromatográfica sería trabajar a temperatura ambiente (25-30°C) y observar cómo varían los tiempos de elución y la resolución de los analitos al incrementar la temperatura (p. ej.: de 5°C en 5°C) y el flujo de la fase móvil (p.ej.: de 0,1mL/min en 0,1mL/min).
Selección de la fase estacionariaLa selección del tipo de columna depende de la naturaleza físico-química del analito que se pretende separar, del tipo y modalidad de HPLC utilizadas y de cuál sea la finalidad de la separación cromatográfica.
Para la correcta selección de una columna analítica deberían considerarse los siguientes aspectos:
1. Selección del tamaño de poro. La selección del tamaño de poro dependerá de la masa molar del analito. Debe seleccionarse un relleno de columna con un tamaño de poro comprendido entre 60Å y 150Å si la masa molar del analito es inferior a, aproximadamente, 2.000g/mol. En caso contrario, debe utilizarse un relleno de columna con un tamaño de poro entre 200Å y 300Å.
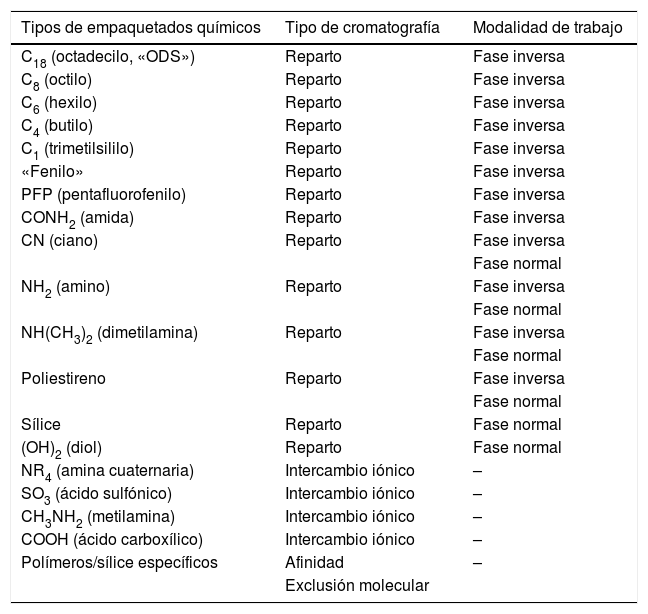
2. Selección del empaquetado químico. En la actualidad, existe una gran variedad de columnas comerciales disponibles con diferentes tipos de empaquetados químicos (tabla 2). En la HPLC de reparto en fase inversa, un buen punto de partida podría ser emplear empaquetados químicos del tipo C18 o C8. Si los analitos de la muestra no se separan adecuadamente, se deberían utilizar las columnas de CN y fenilo. Estas pueden ofrecer diferencias significativas en selectividad frente a las fases alquílicas lineales a la hora de efectuar la separación. En general, los analitos con elevada masa molar, como las proteínas, se separan mejor en columnas de fase inversa con empaquetados químicos de cadena corta (C3, CN) y los péptidos y los analitos con baja-media masa molar, se separan mejor en columnas con empaquetados químicos de cadenas más largas (C8, C18).
Principales empaquetados químicos empleados en las columnas analíticas y el tipo de cromatografía y modalidad de trabajo en la que suelen utilizarse
| Tipos de empaquetados químicos | Tipo de cromatografía | Modalidad de trabajo |
|---|---|---|
| C18 (octadecilo, «ODS») | Reparto | Fase inversa |
| C8 (octilo) | Reparto | Fase inversa |
| C6 (hexilo) | Reparto | Fase inversa |
| C4 (butilo) | Reparto | Fase inversa |
| C1 (trimetilsililo) | Reparto | Fase inversa |
| «Fenilo» | Reparto | Fase inversa |
| PFP (pentafluorofenilo) | Reparto | Fase inversa |
| CONH2 (amida) | Reparto | Fase inversa |
| CN (ciano) | Reparto | Fase inversa |
| Fase normal | ||
| NH2 (amino) | Reparto | Fase inversa |
| Fase normal | ||
| NH(CH3)2 (dimetilamina) | Reparto | Fase inversa |
| Fase normal | ||
| Poliestireno | Reparto | Fase inversa |
| Fase normal | ||
| Sílice | Reparto | Fase normal |
| (OH)2 (diol) | Reparto | Fase normal |
| NR4 (amina cuaternaria) | Intercambio iónico | – |
| SO3 (ácido sulfónico) | Intercambio iónico | – |
| CH3NH2 (metilamina) | Intercambio iónico | – |
| COOH (ácido carboxílico) | Intercambio iónico | – |
| Polímeros/sílice específicos | Afinidad | – |
| Exclusión molecular |
3. Selección del tamaño de partícula. El tamaño de partícula estándar es de 5μm aunque también pueden utilizarse tamaños entre 1,3μm y 3,0μm. Si es necesario realizar una separación cromatográfica más rápida o con una mayor resolución y eficacia es preferible utilizar columnas con un tamaño de partícula de 3,0μm o menor. Cabe destacar que las columnas con un tamaño de partícula menor de 3,0μm operan a una presión<300bar y pueden utilizarse en la mayoría de cromatógrafos líquidos. En cambio, si el tamaño de partícula de la columna es<2,0μm, se alcanzan presiones ≥ 400bar y se precisan cromatógrafos líquidos que permitan soportar estas elevadas presiones.
4. Diámetro interno de la columna. El diámetro interno de la columna define cuánto material (masa o volumen) se puede inyectar en la columna. Cuanto menor sea el diámetro, menor será la cantidad de material que se pueda inyectar manteniendo una aceptable resolución. Las columnas más empleadas presentan un diámetro interno de 4,6mm. Cabe destacar que las columnas con un diámetro interno menor proporcionan una mayor capacidad de detección y, por consiguiente, suelen utilizarse en la HPLC acoplada a la espectrometría de masas o en procedimientos cromatográficos desarrollados con un volumen de muestra limitado.
5. Longitud de la columna. Las columnas más utilizadas presentan una longitud de 100mm, 150mm o 250mm conjuntamente con un tamaño de partícula de 5μm. Estas condiciones proporcionan tiempos de cromatografía comprendidos entre 20-30min. Si se desea acortar el tiempo de cromatografía, se deben emplear columnas con una longitud menor (30mm, 50mm o 75mm) pero con un tamaño de partícula ≤ 3,5μm.
Preparación de soluciones y materiales diversosReactivos y disolventesUn aspecto esencial cuando se trabaja con procedimientos cromatográficos es que los reactivos genéricos y disolventes que se emplean para preparar la fase móvil, los materiales de calibración o de control, los estándares internos y otras soluciones, presenten una elevada pureza (calidad HPLC o HPLC-MS). De no ser así, se puede comprometer la medición de la magnitud en estudio (separación, resolución y eficacias cromatográficas inapropiadas que pueden dar lugar, principalmente, a una menor reproducibilidad y capacidad de detección), así como condicionar el buen funcionamiento del sistema cromatográfico (obstrucción de cánulas, fritas, tubos, válvulas, inyectores, detectores, etc.).
Materiales de calibración y materiales de controlSiempre que sea posible, es recomendable que se utilicen materiales de calibración y de control comerciales y que su preparación se realice siguiendo las instrucciones y recomendaciones descritas por el fabricante. Por otra parte, los materiales de calibración deben presentar valores trazables a una referencia adecuada25. Desafortunadamente, son pocas las empresas del diagnóstico in vitro que fabrican y disponen de este tipo de materiales para sistemas cromatográficos por lo que, en estos casos, los laboratorios clínicos deberán preparar sus propios materiales de calibración y de control.
PreparaciónSi el laboratorio ha de preparar los materiales de calibración y control, debe utilizar13,15,22,23,25:
- -
Un material de pureza conocida y que, si es posible, sea trazable a una referencia de la mayor calidad metrológica posible.
- -
Una mezcla del líquido biológico en estudio (plasma, suero, orina, etc.) pero que no contenga el analito en cuestión (material de blanco).
Cabe destacar que si el analito es un componente biológico, como material de blanco se podría utilizar una solución de cloruro de sodio 154mmol/l y albúmina 50g/l, una solución salina comercial o agua calidad HPLC o HPLC-MS, siempre y cuando se conozca que el efecto matriz es mínimo o despreciable. Si existe un efecto matriz significativo, es recomendable utilizar una mezcla del líquido biológico (plasma, suero, orina, etc.) y aplicar un procedimiento que permita compensar el efecto matriz existente, por ejemplo, mediante el procedimiento de la adición estándar.
El número mínimo recomendable de materiales de calibración a preparar es de seis (sin contar los materiales de blanco o calibradores 0) y con valores que cubran el intervalo de medida en estudio.
El número mínimo recomendable de materiales de control a preparar es de tres. Dos de los materiales deben presentar valores por debajo («control bajo») y por encima («control alto») del intervalo de referencia, intervalo terapéutico o valor de decisión clínica (valor discriminante), y el tercer material debe presentar un valor intermedio o cercano al valor discriminante («control medio»). Además, el «control bajo» debe presentar un valor aproximadamente tres veces el del límite de cuantificación.
Para minimizar errores, es recomendable que el procedimiento de preparación de los materiales de calibración y de control se realice de la siguiente manera:
- 1)
Preparar el material de blanco a partir de una mezcla de muestras de pacientes presuntamente sanos (se recomienda utilizar un mínimo de 10 muestras). Una vez realizada la mezcla, debe verificarse que en la misma no existe el analito. Esta verificación se puede llevar a cabo utilizando la información clínica de cada uno de los pacientes empleados así como, procesando la muestra en el sistema cromatográfico.
- 2)
Preparar dos soluciones primarias del analito, una para los materiales de calibración y otra para los materiales de control, en un disolvente apropiado (agua, metanol, acetonitrilo, dimetisulfóxido, entre otros, dependiendo de la solubilidad del analito). Es necesario que las dos soluciones se preparen a partir de pesadas diferentes.
- 3)
Preparar diferentes soluciones de trabajo (seis para los materiales de calibración y tres para los materiales de control) a partir de la correspondiente solución primaria del analito y del material de blanco. En algunos casos, debido a que los diferentes disolventes orgánicos utilizados en la preparación de la solución primaria pueden provocar la precipitación del material de blanco empleado, es preferible realizar distintas soluciones secundarias (seis para los materiales de calibración y tres para los materiales de control), en agua calidad HPLC o HPLC-MS, y preparar las soluciones de trabajo a partir de estas soluciones secundarias y el material de blanco.
Siempre que sea posible, deberían prepararse nuevas soluciones de trabajo para los materiales de calibración y de control cada vez que se realice la medición de la magnitud en estudio, a menos que se pueda garantizar que estas son estables en unas condiciones de almacenamiento determinadas (p. ej.: a -80°C).
Valores asignados. Trazabilidad e incertidumbre de medidaSi el laboratorio clínico prepara los materiales de calibración, es responsable de asegurar la trazabilidad metrológica de los valores asignados hasta las referencias de mayor jerarquía metrológica asequibles y de calcular la incertidumbre de los valores asignados. Para ello, debe usar la información relacionada con su preparación. Además, la preparación debe realizarse utilizando instrumentos de medida (pipetas, matraces aforados, balanzas analíticas, entre otros) adecuadamente calibrados.
La Comisión de Metrología y Sistemas Analíticos dispone de recomendaciones relacionadas con la utilización de calibradores con trazabilidad metrológica25, la calibración de instrumentos de medida26,27 y de cómo debe estimarse la incertidumbre de medida28.
En el Material suplementario 1 se muestra un ejemplo práctico de cómo realizar la preparación de los materiales de calibración y de control, el cálculo de la incertidumbre de medida de los valores asignados a los mismos, así como la información relativa a la trazabilidad de los resultados de medida.
Soluciones de patrones internosLa utilización de patrones internos es una práctica común en HPLC cuando se utilizan los sistemas cromatográficos con una finalidad cuantitativa22–24. Un patrón interno es una sustancia que se añade a todas las muestras, materiales de calibración y materiales de control en una cantidad conocida constante, permitiendo compensar o reducir algunos errores que se puedan producir en el procedimiento de medida (p. ej.: errores atribuibles a la preparación de la muestra) o bien que puedan afectar al sistema cromatográfico (errores en el pipeteo de muestras por parte del muestreador automático, compensación de fluctuaciones en la señal analítica que puedan producirse en el detector, compensación de un posible efecto matriz, entre otros).
Idealmente, un patrón interno debería cumplir las siguientes características13–15,22,23:
- -
No encontrarse en la muestra a estudiar.
- -
Presentar una composición y estructura química similar a la del analito. En la HPLC-MS o HPLC-MS/MS, siempre que sea posible, debería utilizarse el propio analito marcado con isótopos estables (2H, 13C, 15N, entre otros).
- -
Presentar una elevada pureza. Además, se deberían conocer cuáles son sus impurezas.
- -
Ser suficientemente estable en solución como para garantizar que no se degradará durante los procesos de preparación de la muestra y de separación cromatográfica.
- -
Proporcionar una señal suficiente en el detector cromatográfico.
- -
Presentar una resolución cromatográfica claramente diferenciada del analito. Esta característica no es necesaria si se emplea un espectrómetro de masas como detector cromatográfico.
- -
Eluir después del analito, de tal manera que permita confirmar que la separación cromatográfica se ha producido correctamente. Si se utiliza un espectrómetro de masas como detector cromatográfico, es suficiente —incluso necesario la mayoría de las veces— que el analito eluya al mismo tiempo de retención que el patrón interno.
Siempre que sea posible, es recomendable que se empleen patrones internos comerciales y que su preparación se realice siguiendo las instrucciones y recomendaciones descritas por el fabricante. En caso contrario, su preparación puede realizarse de la siguiente manera13–15,22,23:
- 1)
Preparar una solución primaria de patrón interno en un disolvente apropiado (agua, metanol, acetonitrilo, dimetilsulfóxido, entre otros, dependiendo de la solubilidad del mismo).
- 2)
Preparar, a partir de la solución primaria, una solución de trabajo de patrón interno, en un disolvente apropiado, que produzca una señal analítica suficiente y estable en el detector cromatográfico. Es recomendable preparar una solución de trabajo a una concentración similar a la que tendría el analito a valores fisiológicos, dentro del intervalo de referencia o terapéutico o cercanos al valor discriminante.
Siempre que sea posible, se debería preparar una solución de trabajo de patrón interno nueva cada vez que se realice la medición de la magnitud en estudio, a no ser que se pueda garantizar que esta es estable en unas condiciones de almacenamiento determinadas (p. ej.: a -80°C).
En el Material suplementario 2 se muestra un ejemplo práctico de cómo llevar a la selección de un patrón interno y cómo realizar la preparación de la solución de trabajo del patrón interno.
CalibraciónLos principales procedimientos de calibración empleados en la HPLC son los del patrón externo, del patrón interno (el más utilizado), de normalización de áreas y de la adición estándar22–24.
El proceso de calibración y la obtención de la curva de calibración está sujeto a una serie de condiciones13–15:
- -
La curva de calibración debe estar definida por un mínimo de seis materiales de calibración. Adicionalmente, han de prepararse dos materiales de blanco, uno que no contenga ni el analito en estudio ni el estándar interno (si se emplea), y otro que solo contenga el estándar interno (si se utiliza).
- -
El material de calibración que presenta el valor más bajo de la magnitud en estudio (calibrador 1) debe presentar un valor cercano al límite de cuantificación.
- -
El material de calibración que presenta el valor más alto de la magnitud en estudio (calibrador 6) debe coincidir con el límite superior del intervalo de medida previamente seleccionado.
- -
Los materiales de calibración y los materiales de blanco deben procesarse por duplicado.
- -
Siempre que sea posible, la curva de calibración debe estar representada por la ecuación de una línea recta.
- -
Se puede utilizar cualquier algoritmo matemático que permita ajustar la curva de calibración (regresión lineal, polinómica, múltiple, recíproca, logarítmica, exponencial, entre otros).
- -
Los valores obtenidos en los materiales de blanco no deben considerarse durante la realización del ajuste matemático que representará la curva de calibración.
- -
Para cada material de calibración procesado, debe calcularse la diferencia relativa porcentual entre el valor obtenido mediante el ajuste matemático (valor obtenido) y su correspondiente valor asignado dividido por el valor asignado. Esta diferencia debe estar comprendida entre±15%, excepto para el calibrador 1 (calibrador con un valor cercano al límite de cuantificación) donde esta diferencia puede estar comprendida entre±20%. Como mínimo, para el 75% de los materiales de calibración procesados debe cumplirse esta condición. Además, entre los duplicados de un mismo material de calibración, al menos uno de ellos debe cumplir este requisito.
Si alguno de los materiales de calibración no cumple estas condiciones, el valor obtenido para el mismo debe eliminarse, volver a realizar el ajuste de la curva de calibración, calcular de nuevo las distintas diferencias relativas porcentuales y comprobar el cumplimiento de dichas condiciones.
Validación del procedimiento de medida desarrolladoUna vez desarrollado el procedimiento cromatográfico, el laboratorio clínico debe llevar a cabo una validación del mismo antes de ser utilizado con muestras de pacientes, con el objeto de confirmar que este será adecuado para sus aplicaciones clínicas29,30.
En el Material suplementario 3, se muestra un ejemplo que contiene los principales aspectos a considerar en el desarrollo de un procedimiento cromatográfico.
