




La enfermedad de Castleman (EC), también denominada hiperplasia linfoide angiofolicular, fue descrita por primera vez en 1956 por Benjamín Castleman1. Es un proceso linfoproliferativo poco frecuente y de etiología no bien conocida que se caracteriza por hiperplasia de los ganglios linfáticos. Existen 3 variedades histológicas bien diferenciadas, la hialino-vascular, la plasmocelular y mixta, que a su vez, pueden ser localizadas o multicéntricas2. La forma hialino-vascular suele ser asintomática y localizada en mediastino, mientras que la plasmocelular se presenta frecuentemente con sintomatología sistémica y puede ser localizada o multicéntrica asociándose con mayor frecuencia a procesos malignos. La edad media de presentación es de 20 años, con un rango etario que va desde los 2 a 85 años siendo estos en estas edades extremas mucho más infrecuentes. Es por ello que presentamos el caso de un paciente octogenario que tras realización de pruebas complementarias y estudio exhaustivo presentó EC tipo plasmocelular difuso que consideramos de interés por lo poco frecuente de esta entidad y sus implicaciones diagnósticas.
Se trata un varón de 86 años con los antecedentes de hipertensión arterial e hipertrigliceridemia y enfermedad de Perthes en cadera izquierda. Respecto a su situación basal previa, mantenía independencia para las actividades básicas e instrumentales de la vida diaria y mantenía la deambulación autónoma con ayuda de un bastón. No presentaba déficit amnésico ni alteraciones emocionales ni sensoriales. Era soltero y vivía en su domicilio bajo la supervisión estrecha de sobrinos. Acudió al hospital por cuadro de un mes de evolución consistente en astenia y anorexia junto con pérdida de peso objetiva de 4kg en el último mes. Además se acompañaba de dolor abdominal difuso sobre todo en epigastrio e irradiado en hipocondrio derecho sin síndrome febril asociado, por lo que se decide su ingreso en planta de Geriatría para estudio.
A la exploración solo se objetivó abdomen levemente distendido con dolor difuso a la palpación profunda con hepatomegalia y esplenomegalia palpables, sin datos de irritación peritoneal y sin palparse adenopatías patológicas.
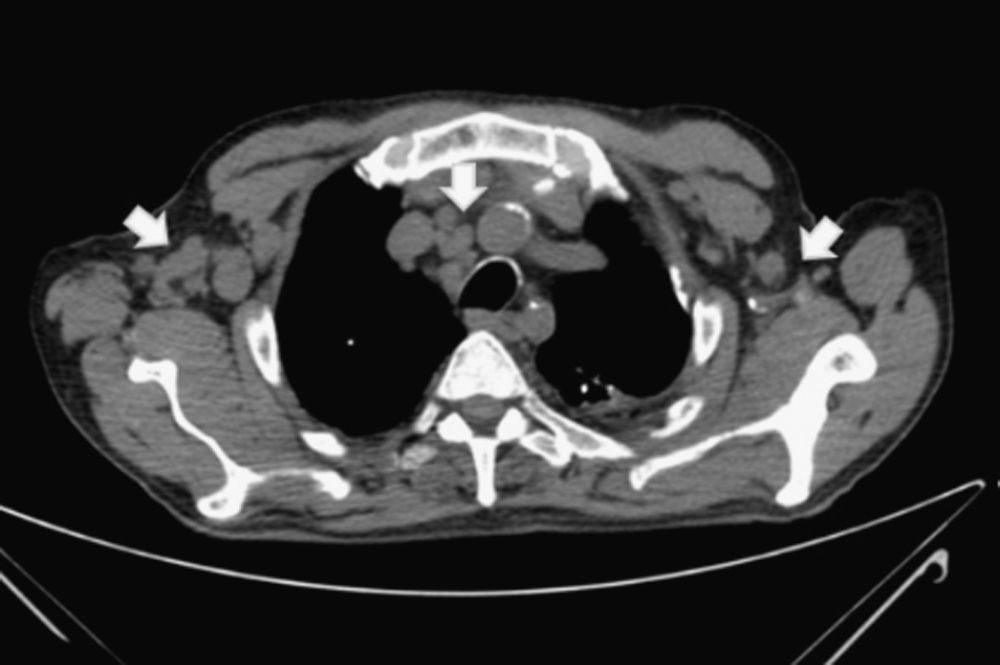
En la analítica al ingreso se encontraron los valores siguientes: leucocitos 10,6 miles/μl (neutrófilos 30%, linfocitos 51%, monocitos 16%), Hb 10,9g/dl, hematocrito 32%, plaquetas 110.000 miles/μl. VSG 104mm/h, glucosa 114g/dl, urea 65mg/dl, creatinina 1,8mg/dl, proteínas 7,6g/dl, albúmina 2,7g/dl. Perfil tiroideo, lipídico, B12, fólico y marcadores tumorales dentro de la normalidad. Las serologías para virus de hepatitis B y C, Epstein-Barr, citomegalovirus, toxoplasma y virus de la inmunodeficiencia humana resultaron negativas. El factor reumatoide y los anticuerpos antinucleares, así como el Mantoux fueron negativos. En el proteinograma se encontró un pico monoclonal en región gamma, IgG lambda y en orina de 24h cadena ligera Kappa. En la gammagrafía ósea se encontraron datos compatibles con poliartritis y en la serie ósea imagen compatible con lesión osteolítica de grado IB a nivel occipital. Por último, en la tomografía computarizada (TC) toracoabdominal se hallaron resultados sugerentes de síndrome linfoproliferativo con adenopatías múltiples, sobre todo a nivel axilar, paraaórticas y retroperitoneales de más de 15mm (fig. 1). Se realizó el aspirado y la biopsia de médula ósea no mostrando infiltración. Ante la falta de diagnóstico se decidió exéresis de una adenopatía axilar accesible. El estudio microscópico anatomopatológico reveló una pérdida parcial de la arquitectura con hiperplasia linfoide focal marcada y expansión de la zona interfolicular ocupada por abundantes plasmocitos compatible con la variedad plasmocelular.

Tras este diagnóstico se continuó con hidratación adecuada para evitar mayor deterioro de la función renal, control del dolor con pirazolonas y paracetamol junto con tratamiento con bifosfonatos y suplementos orales de calcio y vitamina D. Con respecto al tratamiento específico para la EC y el mieloma, se decidió iniciar tratamiento oral con esteroides a dosis altas, junto con melfalán a dosis de 0,15mg/kg durante 7 días a la espera de evolución y respuesta clínica.
Fue dado de alta del servicio de Geriatría, con sintomatología controlada en situación de dependencia funcional reversible en el contexto de ingreso prolongado y, probablemente, por la toma de corticoterapia sistémica, iniciándose en planta programa de rehabilitación y reeducación de la marcha, que se continuó al alta de forma ambulatoria.
Hemos presentado el caso de un paciente octogenario afecto de EC asociado además a mieloma múltiple con lesión en región occipital. La franja etaria es amplia pero infrecuente en este grupo de edad, de ahí lo interesante del caso.
La EC es una entidad rara, de etiopatogenia desconocida, en probable relación con el herpes virus tipo 8, fallo en la inmunorregulación, expresión aumentada del gen codificador de interleucina-6 y aumento de la producción por parte de los nódulos hiperplásicos3.
La localización más frecuente es la mediastínica y la abdominal siendo rara la afectación cervical (10%)4. La forma histológica más frecuente de la EC es la hialino-vascular que afecta a individuos jóvenes, suele manifestarse como una adenopatía única bien delimitada asintomática o con escasa afectación sistémica (astenia, febrícula) y presenta un curso benigno y curable mediante exéresis quirúrgica del ganglio.
Por el contrario, en la variedad plasmocelular, el tejido interfolicular está compuesto por sábanas de células plasmocíticas con escaso estroma vascular. Se suele presentar en la edad adulta, y clínicamente se caracteriza por fiebre, disminución de peso, astenia, linfadenopatías generalizadas, hepatoesplenomegalia, lesiones cutáneas y neuropatía. En los estudios de laboratorio destaca una elevación de la velocidad de sedimentación, hipergammaglobulinemia y anemia de trastornos crónicos. Las manifestaciones sistémicas se han asociado a un aumento de producción de IL-6 por las células plasmocelulares. El curso es agresivo y a menudo fatal, con una media de supervivencia de 30 meses5, generalmente debido al desarrollo de complicaciones infecciosas y neoplasias, particularmente sarcoma de Kaposi, linfomas no Hodking y plamocitomas6,7. Las técnicas de imagen como la TC o la resonancia magnética nuclear nos ayudan al diagnóstico informándonos de la localización, número y tamaño de las adenopatías afectadas, sobre todo teniendo en cuenta que la localización más frecuente es a nivel de mediastino. Pero el diagnóstico definitivo lo obtendremos mediante la biopsia del ganglio8,9.
El tratamiento de la forma localizada es la resección quirúrgica lo más radical posible, tanto en el tipo hialinovascular como el tipo plasmocelular, con una curación de casi el 100% de los casos y con excepcionales recurrencias. Otras terapias como la resección parcial y la radioterapia, se reservan para los pacientes con lesiones no resecables. En la forma multicéntrica, es difícil hablar del tratamiento ideal, dado el escaso número de pacientes de las series publicadas y la amplia variabilidad de los resultados descritos en la literatura. Se han utilizado tratamientos muy diversos como los esteroides en monoterapia o conjuntamente con agentes quimioterápicos (ciclofosfamida, vincristina, clorambucilo), sin evidencia de eficacia de ninguno de ellos, y con respuestas clínicas muy variadas10. El seguimiento a largo plazo y la vigilancia periódica aún no están definidos para ninguna de las formas clínicas, aunque tal vez sea lo más prudente para detectar recurrencias de la enfermedad o el desarrollo de enfermedades malignas.
