




Se expone el caso de una paciente de 73 años que presentó cuadro constitucional y dolor en miembros inferiores, con sospecha inicial de mieloma múltiple. Durante el proceso diagnóstico aparecieron discordancias entre la evolución clínica y los hallazgos en pruebas complementarias. Tras un curso clínico fulminante en pocos días, la paciente falleció; se obtuvo el diagnóstico post mórtem de linfoma angioinmunoblástico. Se repasan los aspectos fundamentales de esta patología poco frecuente, de patogenia todavía incierta.
We describe the case of a 73-year-old woman with constitutional disorder and pain in the lower limbs, leading to initial suspicion of multiple myeloma. During the diagnostic process, there were discrepancies between the clinical course and findings of complementary tests. After a fulminant clinical course for a few days, the patient died, and a postmortem diagnosis of angioimmunoblastic lymphoma was established. We review the main aspects of this highly infrequent disease, the pathogenesis of which remains uncertain.
Mujer de 73 años que acudió a urgencias por cuadro constitucional de 3 meses de evolución, con pérdida de 6kg de peso en el último mes y dolor sordo en cara anterior tibial de ambos miembros inferiores y escasa respuesta a tratamiento analgésico. Entre sus antecedentes personales destacaban: hipertensión arterial, diabetes mellitus tipo II, hipercolesterolemia, fibrilación auricular paroxística, osteoporosis con aplastamientos de vértebras lumbares L2 y L4 y cirugía de catarata en ojo derecho.
Respecto a su situación basal, la paciente era independiente para las actividades básicas e instrumentales de la vida diaria; presentó deterioro funcional días antes del ingreso hospitalario. Mentalmente presentaba ánimo triste reactivo. En el ámbito social era viuda, con dos hijos y buen apoyo familiar.
En la exploración física no se encontraron datos relevantes y se descartó la presencia de adenopatías y visceromegalias.
En la analítica destacaban una velocidad de sedimentación globular de 103mmHg, discreta anemia normocítica-normocrómica hiperregenerativa, un frotis de sangre periférica con hematíes en pila de monedas y bioquímica con ligera elevación de GOT, GPT y gamma-GT. Las serologías virales resultaron negativas, así como de Toxoplasma, Leishmania y Rickettsia, y el Mantoux.
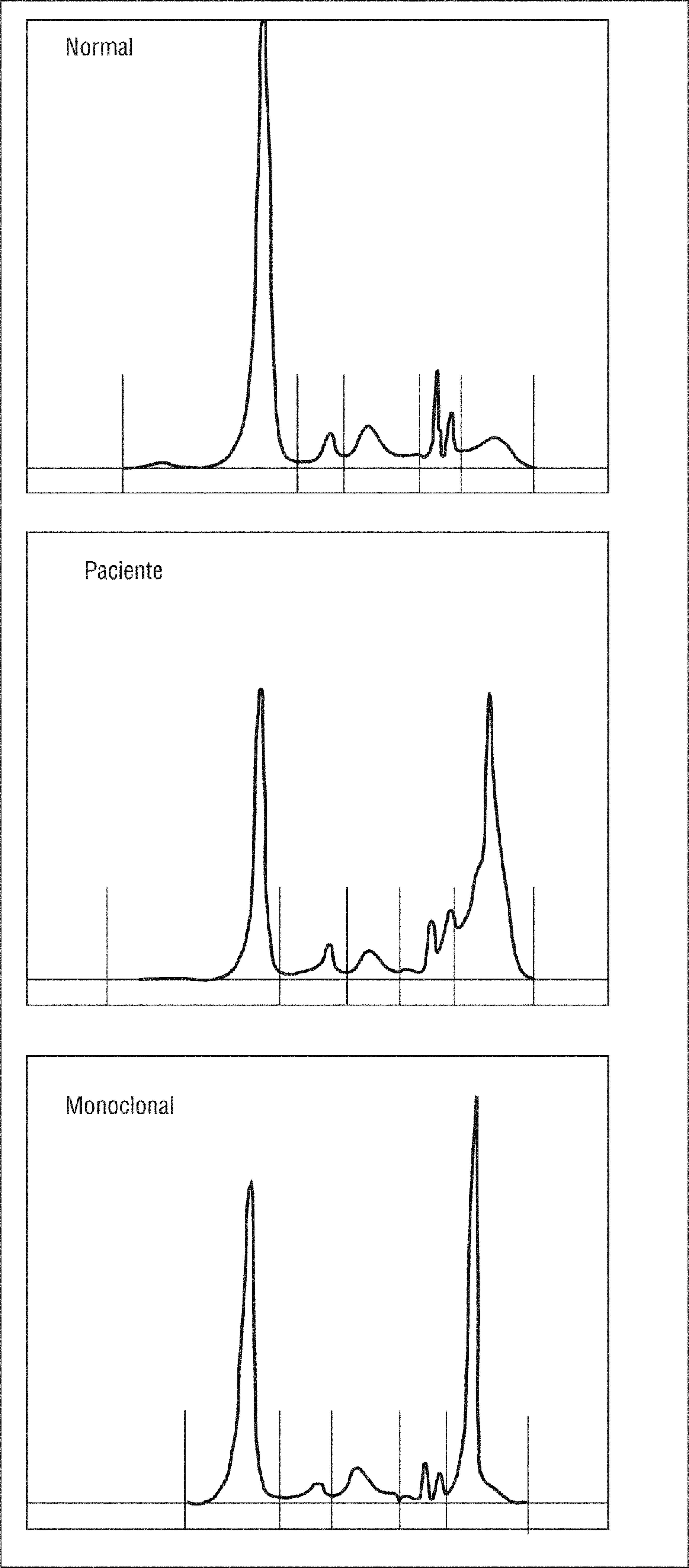
Se observó hiperproteinemia (9,2g/dl) con marcada hipergammaglobulinemia (49,6%). Tras la inmunoelectroforesis y la cuantificación de inmunoglobulinas, se objetivó una gammapatía policlonal con elevación de los niveles de inmunoglobulina (Ig) G (4.580mg/dl), IgA (934mg/dl) e IgM (493mg/dl); el cociente kappa/lambda fue normal. No se demostró paraproteinemia con técnicas de inmunofijación, hallazgo típico del mieloma (fig. 1). Sorprendentemente el aspirado de médula ósea mostró celularidad normal, con plasmacitosis del 36 al 70%, muy sugestivo de mieloma.

En resumen, se trata de una paciente que ingresó con sospecha de mieloma múltiple, confirmado por aspirado medular, en la que, sin embargo, aparecen datos de gammapatía policlonal en la inmunoelectroforesis. Ante estos hallazgos, se pensó como entidades diagnósticas la posibilidad de enfermedad de Castleman, linfoma con alta carga tumoral y como posibilidad más rara un mieloma no secretor.
A los 20 días del ingreso hospitalario, la paciente presentó bruscamente fiebre elevada, lesiones cutáneas purpúricas y adenopatías dolorosas con hepatoesplenomegalia; falleció 2 dos días después.
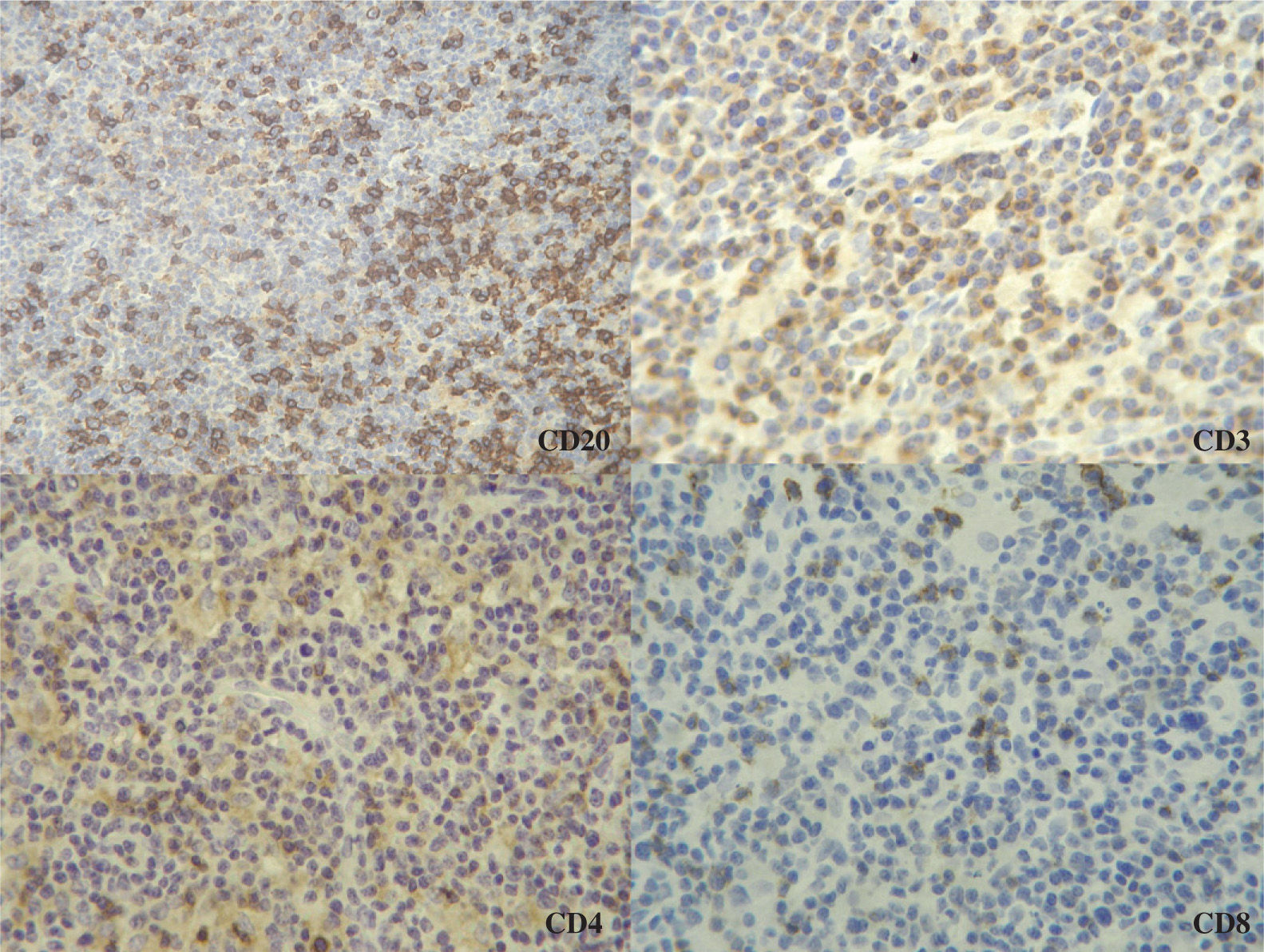
La necropsia solicitada se informó como linfoma T periférico CD4+ asociado al virus de Epstein-Barr, y plasmocitosis policlonal, con afectación ganglionar generalizada, hepatoesplénica, medular, del anillo de Waldeyer, mamaria, pulmonar bilateral y cutánea, compatible con linfoma angioinmunoblástico. En los ganglios (fig. 2) existía infiltración neoplásica de crecimiento difuso, de citología polimorfa, con expresión de antígenos T, frecuentes acúmulos de células plasmáticas policlonales, ocasionales células CD30+ y abundantes células EBER+(ARN específico de la fase latente del virus de Epstein-Barr), todo ello compatible con un linfoma T-angioinmunoblástico (T-AIL). A diferencia de casos paradigmáticos, las células dendríticas foliculares y los linfocitos B eran muy escasos.
DISCUSION y T (CD3, CD4, CD8).")
El interés del caso se centra en que el linfoma angioinmunoblástico, a pesar de presentarse en ancianos, es una entidad poco frecuente, que en este caso cursó con agresividad y rapidez; los síntomas iniciales son anodinos, asociados únicamente con deterioro funcional subagudo. Las pruebas de laboratorio iniciales orientaron hacia el diagnóstico de mieloma, debido a la hiperproteinemia, la velocidad de sedimentación elevada, la anemia normocítica y el aspirado medular compatible. En pocos días el deterioro clínico e inmunológico de la paciente fue fulminante, con fracaso multiorgánico fatal, por lo que el diagnóstico se realizó postmortem.
Los linfomas T-AIL representan el 2% de los linfomas no hodgkiniano y el séptimo tipo de cáncer en frecuencia en el Reino Unido1. Es una enfermedad de predominio en ancianos y presenta su máxima incidencia hacia la sexta o séptima décadas de la vida2.
No se han descrito factores de riesgo ni agentes etiológicos consistentes hasta el momento. Se ha asociado con linfoma T-angioinmunoblástico una serie de enfermedades infecciosas, como la tuberculosis, criptococosis, o algunos virus linfotropos, como el virus de Epstein-Barr, el virus de la inmunodeficiencia humana, virus de la hepatitis C3, el herpes virus humano tipo 64 y tipo 85; el primero de ellos es el más significativo. El virus de Epstein-Barr aparece abundantemente en los linfocitos T del 95% de los pacientes con linfoma T angioinmunoblástico6, aunque existen otros estudios con inmunohistoquímica e hibridación in situ que muestran que las células infectadas por el virus de Epstein-Barr serían los linfocitos B7.
El linfoma T angioinmunoblástico suele presentarse como una enfermedad sistémica, con poliadenopatías generalizadas y síntomas B, y simula con frecuencia un proceso infeccioso. Como consecuencia de la disfunción inmunológica subyacente, suelen aparecer síntomas inflamatorios sistémicos, infecciones y fenómenos autoinmunes. Los hallazgos de laboratorio más frecuentes son anemia, hipergammaglobulinemia, autoanticuerpos y velocidad de sedimentación elevada.
El diagnóstico diferencial del linfoma angioinmunoblástico incluye procesos inflamatorios y neoplásicos. Los hallazgos en sangre periférica y médula ósea son inespecíficos.
Para el diagnóstico es indispensable la realización de una biopsia ganglionar de uno de los ganglios de mayor tamaño; se obtiene información morfológica, consistente en el borramiento parcial de la arquitectura ganglionar por un infiltrado celular polimorfo. En algunos pacientes los hallazgos histológicos podrían solaparse con linfadenitis complejas, enfermedad de Castleman multicéntrica, linfomas difusos de células grandes B y linfomas Hodgkin2.
Las localizaciones extraganglionares más frecuentes son médula ósea, bazo, piel y pulmón8. Las características histológicas de la afectación extraganglionar no son específicas, aunque suelen existir paralelismos con los infiltrados ganglionares.
El pronóstico de los pacientes con linfoma angioinmunoblástico es infausto, con una supervivencia media inferior a los 36 meses9. La mayoría de los pacientes fallece por complicaciones infecciosas y no por la propia neoplasia.
