



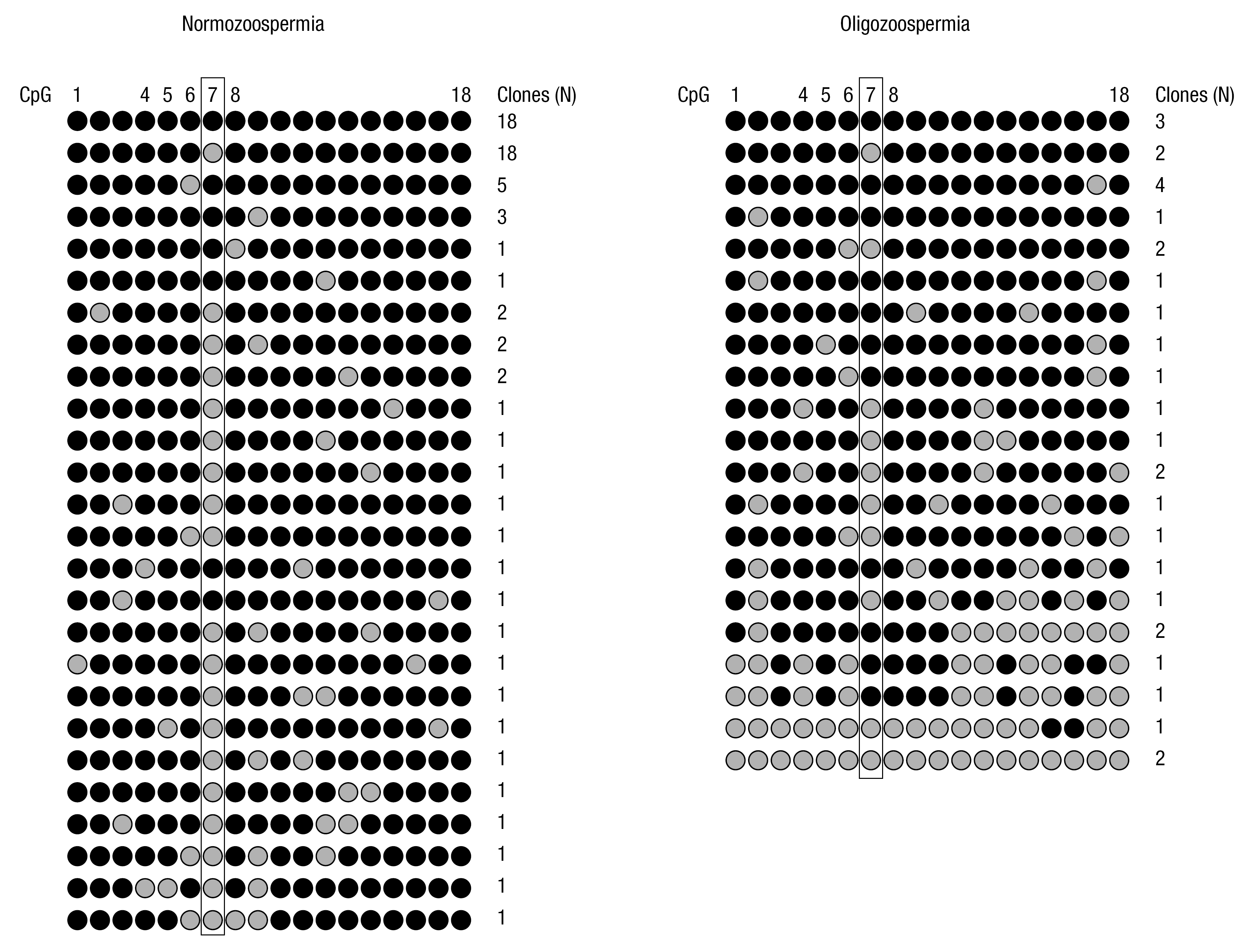
INTRODUÇÃO
O imprinting genómico é um mecanismo de regulação da expressão dos genes que permite apenas a expressão de um dos alelos parentais. Assim, e ao contrário da maioria dos genes em que a expressão é bi-alélica, os genes que estão submetidos a este mecanismo têm uma expressão mono-alélica. De um modo geral, os genes submetidos a imprinting encontram-se próximos ou associados no genoma (domínios de imprinting). Os domínios de imprinting mais estudados, situam-se nos cromossomas 11 e 15. Na região 11p15.5 encontram-se os genes H19 e IGF2, cujo défice de imprinting origina o síndrome de Beckwith-Wiedemann. Na região 15q11-13 situam-se os genes SNRPN e UBE3A, cujo défice de imprinting origina o síndrome de Prader-Willi e o síndrome de Angelman. A expressão aberrante destes genes sujeitos a imprinting pode provocar defeitos no desenvolvimento embrionário e da placenta, anomalias neurológicas e cancro1.
Os síndromes de Angelman e de Beckwith-Wiedemann são muito raros, estando a sua frequência ligeiramente aumentada nas crianças nascidas por reprodução medicamente assistida, quer por fecundação in vitro (FIV) do espatozóide, por microinjecção intracitoplasmática (ICSI). Existem 3 casos de crianças nascidas por ICSI e com Síndrome de Angelman, e 20 casos de crianças com o Síndrome de Beckwith-Wiedemann nascidas por ICSI (10 casos) e por FIV (10 casos)2. Estes dados sugerem a possibilidade de a transmissão de defeitos de imprinting estar associada às técnicas de reprodução medicamente assistida. Em todas estas crianças acima descritas, a causa do síndrome deveu-se a defeitos na metilação do alelo materno, provavelmente por anomalias na reprogramação genética que ocorre nos embriões em cultura após a fase de 4 células. Em relação à possibilidade da transmissão de defeitos de imprinting no alelo paterno durante a microinjecção por infertilidade masculina, poucos estudos foram efectuados e nenhum se revelou conclusivo3-6.
De um modo geral, a marca de imprinting consiste na metilação das citosinas dos dinucleótidos CG, situados em ilhas CpG, que são zonas do genoma em que a frequência deste dinucleótido é, comparativamente ao resto do DNA, muito elevada. As ilhas CpG encontram-se normalmente associadas ao promotor de um gene. De um modo geral, a metilação de uma ilha CpG inactiva a expressão desse gene. Na maioria dos casos, estas alterações epigenéticas ocorrem apenas num dos alelos do gene, enquanto que o outro alelo permanece desmetilado e activo. Deste modo, o encontro dos dois alelos parentais, um metilado e outro não metilado, paterno e materno, na fertilização, permite um desenvolvimento embrionário normal7. As marcas de imprinting herdadas (oriundas dos espermatozóides e dos ovócitos) são primeiro apagadas (desmetilação global do genoma) nas células germinais primordiais durante as primeiras fases da vida fetal. De seguida, as marcas do imprinting são restabelecidas (metilação preferencial de determinados genes) durante a gametogénese consoante o sexo do feto, ainda durante a vida fetal no caso da ovogénese, e após a puberdade no caso da espermatogénese. Existem duas fases de reprogramação epigenética. A primeira fase é caracterizada por uma desmetilação global de todo o genoma nas células germinais primordiais, fenómeno secundado por uma vaga de metilação dependente do sexo embrionário (imprinting parental) que se efectua nas fases iniciais da gametogénese. A segunda fase ocorre durante o período de desenvolvimento embrionário pré-implantação, verificando-se uma desmetilação dos genomas materno e paterno, com excepção dos genes submetidos a imprinting, que se encontram protegidos deste fenómeno. A maioria dos genes submetidos a imprinting parental são genes essenciais para o desenvolvimento normal do embrião8.
Dos genes sujeitos a imprinting parental, o gene H19 é dos melhores estudados. Nos humanos, localiza-se no cromossoma 11 (11p11.5) e encontra-se metilado no alelo paterno (silenciamento) e não-metilado no alelo materno (transcrição activa). Codifica para um hnRNA que, após processamento (splicing), se transforma num mRNA poli-adenilado com 2,5 Kb. Este mRNA é transportado para o citoplasma, não sendo traduzido em proteína9,10. O gene H19 é essencialmente transcrito nos tecidos mesodérmicos e endodérmicos fetais, sendo inibida a sua transcrição em todos os orgãos após o nascimento, com excepção do fígado11. A desregulação deste sistema está associado com o desenvolvimento de neoplasias fetais, pelo que o mRNA transcrito do gene H19 parece actuar como factor oncofetal12.
Como acontece com a maioria dos genes sujeitos a imprinting, o gene H19 possui uma região diferencialmente metilada (DMR), que se localiza de (2,0) a (3,4) Kb do local de iniciação da transcrição. Esta DMR possui todos os critérios para se considerar que está sujeita a imprinting: a) apresenta metilação paterna específica nas células somáticas; b) está metilada nos espermatozóides, e c) a metilação permanece estável durante o desenvolvimento embrionário pré-implantação13. A DMR do H19 é constituída por 3,5 repetições directas de uma sequência com 400 bp. A sequência com 400 bp possui seis sequências conservadas (hcs 1-6), cada uma com 39 bp. Destas, as hcs 4-6 apresentam homologia com os ratinhos e os ratos5. Dentro de cada hcs existe um número variável de CpGs, das quais um grupo restrito de 4-5 CpGs constitui um núcleo altamente conservado. O núcleo altamente conservado da hcs6, com 5 CpGs, representa um local de ligação (local CTCF6) da proteína CTCF, a qual tem uma função reguladora e limita a actividade dos activadores de transcrição do gene14.
O gene IGF2 localiza-se a (90) Kb do gene H19 e codifica para o factor de crescimento tipo II da insulina (IGF2). Nos ratinhos, verificou-se que a proteína IGF2 tem uma acção mitogénica, funcionando como factor de proliferação e diferenciação celulares, promovendo assim o crescimento embrionário e fetal15. Nos humanos, a expressão da proteína IGF2 é contínua, embora diminua após o nascimento16,17. Os genes H19 e IGF2 apresentam um imprinting oposto, ou seja, o gene IGF2 não sofre metilação diferencial nos gâmetas, sendo a sua expressão monoparental regulada indirectamente pelo imprinting do gene H19. Este mecanismo é explicado pelo facto dos dois genes partilharem a mesma sequência cis-reguladora (região 3' do gene H19) que contém activadores génicos comuns. No caso do alelo paterno, em que a DMR do gene H19 está metilada, a proteína CTCF não consegue ligar-se às CpGs metiladas, pelo que o gene IGF2 é transcrito (por activação do seu promotor pelos activadores endodérmicos comuns aos dois genes), enquanto que a expressão do gene H19 é inibida. A situação oposta ocorre no alelo materno. Neste caso, a CTCF liga-se à DMR do gene H19 porque as CpG não estão metiladas, sendo o gene H19 transcrito e a expressão do IGF2 silenciada (a ligação da CTCF impede o acesso dos activadores ao promotor do gene IGF2)14,18-20. Ou seja, na linha germinal masculina há silenciamento do H19 e transcrição do IGF2, enquanto que na linha germinal feminina há expressão do H19 e silenciamento do IGF2.
Recentemente21, verificamos existir um défice global da metilação do gene H19 nos espermatozóides dos pacientes com oligozoospermia, em 30% dos casos com oligozoospermia grave (< 5 x 106/ml) e em 17,4% dos casos com oligozoospermia moderada (≥ 5 < 20 x 10/ml), mas não nos espermatozóides dos homens com concentração normal de espermatozóides no ejaculado (≥ 20 x 106/ml). No presente estudo, apresentamos uma análise mais detalhada do padrão de metilação do gene H19. Para isso, em vez de estudar a metilação por amostra total de espermatozóides, efectuamos uma análise por espermatozóide através da técnica da clonagem molecular, comparando a normozoospermia com a oligozoospermia grave.
MATERIAL E MÉTODOS
Isolamento de espermatozóides do ejaculado
Sob consentimento informado, estudaram-se 5 ejaculados de homens com parâmetros seminais normais22 e 2 ejaculados de pacientes com oligozoospermia grave (1.180 x 106/ml e 1,6 x 106/ml). Após liquefacção do sémen, os espermatozóides morfologicamente normais e com motilidade progressiva rápida foram purificados por centrifugação de gradientes (Ixaprep; Medicult, Copenhagen, Denmark) seguido de swim-up.
Extracção do DNA dos espermatozóides
A extracção do DNA dos espermatozóides foi efectuada utilizando o método de lise alcalina, com KOH e DTT23.
Mutação dirigida do DNA pelo bissulfito de sódio
O tratamento com bissulfito de sódio permite o estudo da metilação das citosinas do DNA, pois converte as citosinas não metiladas em uracilo, deixando as citosinas metiladas como citosinas. Deste modo, após amplificação da região e sequenciação do DNA, podemos avaliar o estado de metilação de cada citosina individualmente (tabela 1).

O tratamento pelo bissulfito de sódio foi efectuado por dois métodos alternativos, o de Manning et al6 e pelo reagente comercial CpGenome DNA Modification Kit (Intergen).
Amplificação do DNA
A amplificação do DNA foi efectuada por hemi-nested PCR, obtendo-se um produto de 231 bp do gene H19 com 18 CpGs da DMR e que inclui a região hcs6 onde a CFCT se liga (GenBank accession n.º AF125183; nucleótidos 6005-6326)4. Os produtos de PCR foram separados por electroforese num gel a 2,5% de agarose, e visualizados por fluorescência após coloração do DNA com brometo de etídio.
Clonagem dos produtos de PCR
Os fragmentos de DNA amplificados foram clonados com transformação de bactérias, sendo que cada colónia transformada apenas possui o DNA referente a um só espermatozóide. Para este efeito foi utilizado o TOPO TA Cloning Kit (Invitrogen) com a estirpe DH5α de Escherichia coli. A extracção do DNA plasmídico foi efectuada com recurso ao Kit QIAprep Spin Miniprep (Qiagen). A amplificação do DNA foi efectuada por PCR como acima descrito.
Sequenciação do DNA
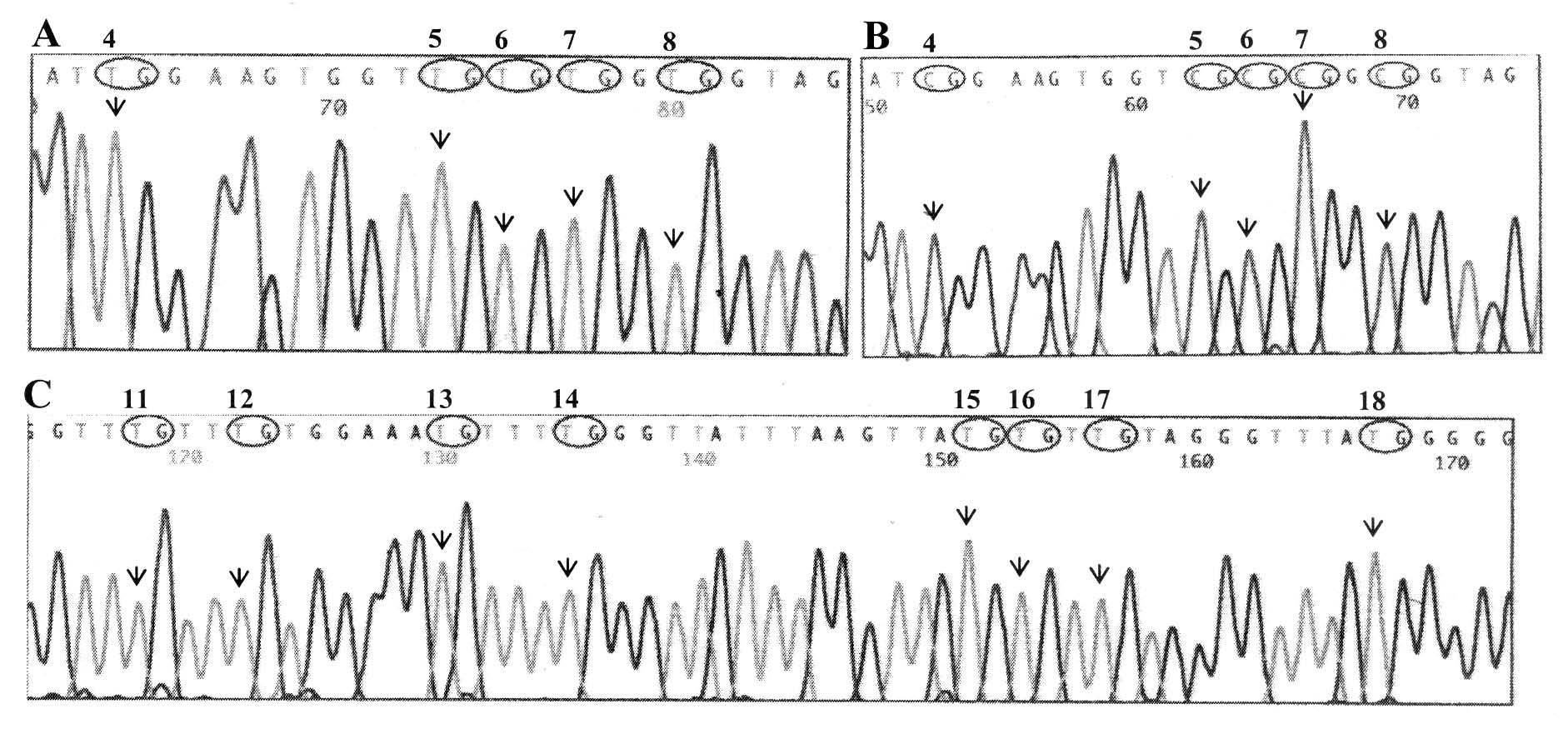
Os produtos de PCR foram purificados com o kit GFX PCR-DNA and Gel Band Purification Kit (Amersham Pharmacia). Após a purificação, os fragmentos de DNA foram sequenciados automaticamente (fig. 1) utilizando o kit BigDye Terminator Cycle Sequencing v1.1 Ready Reaction (Applied Biosystems) num ABI PRISM 310 Genetic Analyzer com software adequado (DNA Sequencing Analysis®, Applied Biosystems).

Figura 1. Análise da metilação após tratamento com o bissulfito de sódio seguido de sequenciação do DNA. A. Exemplo de clone com as CpG 4-8 não metiladas (região de ligação da CTCF). B. Exemplo de clone com as CpG 4-8 metiladas. C. Exemplo de clone com as CpG 11-18 não metiladas.
Análise estatística
As médias entre grupos foram comparadas recorrendo ao teste T de student (2-tailed). A comparação entre grupos relativo às frequências percentuais foi efectuada pelo teste do qui quadrado (2-tailed). Para a análise estatística utilizou-se o programa SPSS 9.0 Windows. O intervalo de confiança foi de 95%, considerando-se o p significativo quando inferior a 0,05.
RESULTADOS
Foram estudados 69 clones obtidos de 5 amostras de sémen de homens com concentração normal de espermatozóides no ejaculado, e 31 clones obtidos de 2 amostras de sémen de pacientes com oligozoospermia grave.
Normozoospermia
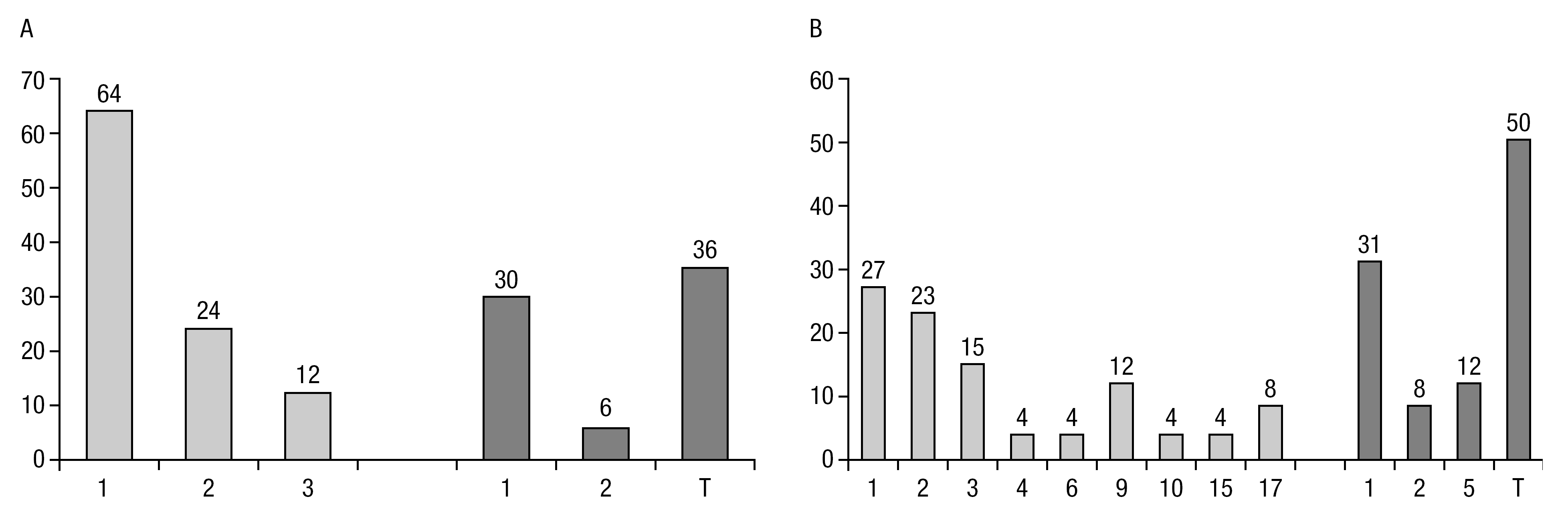
Nos clones derivados das amostras normozoospérmicas (fig. 2), 36/69 (52,2%) dos clones apresentaram um padrão normal de metilação, enquanto que 33/69 (47,8%) dos clones apresentaram metilação incompleta. A CpG na posição 7 representa um local polimórfico (polimorfismo C/T; GenBank acession number AF125183, posição 7966), querendo isto dizer que na população coexistem as formas metilada e não metilada sem qualquer significado patológico: em 39/69 (56,5%) dos clones no total das amostras, em 18/36 (50%) dos clones no grupo restrito da amostra sem défices de metilação noutro local, e em 21/33 (63,6%) dos clones no grupo restrito da amostra com défices de metilação noutro local, o que confirma o carácter polimórfico da CpG-7. Nos 33 clones com alteração da metilação, 21 clones atingiram uma só CG, 8 clones apresentaram não metilação em simultâneo em duas CG, e 4 clones estavam não metilados em três CG em simultâneo (fig. 3A). Nos 33 clones com défice de metilação, o local de ligação da CTCF (CpG 4-8), apenas foi encontrado não metilado parcialmente em 12 clones, 10 clones numa CG e 2 clones em duas CG (fig. 3A). No total dos clones, a média de défices de metilação foi de 0,71 (variação: 0-3; SD: 0,893; SEM: 0,108), e de 1,485 (variação: 1-3; SD: 0,712; SEM: 0,124) considerando apenas os clones com défices de metilação.

Figura 2. Diagrama com o padrão de metilação das 18 CpGs do gene H19 nos clones analisados. A CpG-7 (dentro de rectângulo) é um local polimórfico, pelo que a sua não metilação não corresponde a um défice. O local de ligação da proteína CTCF corresponde às CpG 4-8. Citosina metilada (preto) e citosina não metilada (cinzento).

Figura 3. Frequências percentuais (eixo dos Y) de clones com défice de metilação na (A) normozoospermia (n = 33) e na (B) oligozoospermia severa (n = 26), com o número de CG não metiladas em simultâneo encontrando-se indicado no eixo das abcissas. À esquerda analisa-se a % de clones com 1 ou mais CG não metiladas no total de clones com défice de metilação, e à direita a % de clones com 1 ou mais CG não metiladas na região de ligação da CTCF (CpG4-8).
Oligozoospermia severa
Nos clones derivados das amostras com oligozoospermia severa (fig. 2), 5/31 (16,1%) dos clones apresentaram um padrão normal de metilação, enquanto que 26/31 (83,9%) dos clones apresentaram metilação incompleta. Nos 26 clones com alteração da metilação, 7 clones apresentaram apenas uma CG não metilada, enquanto que os restantes apresentaram várias CG não metiladas em simultâneo: em duas CG (6 clones), três CG (4 clones), quatro CG (1 clone), seis CG (1 clone), nove CG (3 clones), dez CG (1 clone), quinze CG (1 clone) e em dezassete CG (2 clones) em simultaneidade (fig. 3B). De salientar ter-se encontrado um clone com quinze CG não metiladas e dois clones com todas as CG não metiladas. O local de ligação da CTCF (CpG 4-8) foi encontrado não metilado em 13 clones: 8 clones numa só CG, 2 clones em duas CG em simultâneo, e não metilado na sua totalidade em 3 clones. A não metilação polimórfica da CpG-7 foi encontrada em 14/31 (45,2%) dos clones no total das amostras, em 2/5 (40%) dos clones no grupo restrito da amostra sem défices de metilação noutro local, e em 12/26 (46,2%) dos clones no grupo restrito da amostra com défices de metilação noutro local. No total dos clones, a média de défices de metilação foi de 4.097 (variação: 0-17; SD: 4.982; SEM: 0,895), e de 4.885 (variação: 1-17; SD: 5.078; SEM: 0,996) considerando apenas os clones com défices de metilação.
Estudo comparativo
A comparação estatística entre as médias de clones com défices de metilação revelou diferenças significativas entre a normozoospermia e a oligozoospermia grave, quer em relação ao total dos clones (p = 0,001), quer em relação aos clones com défices de metilação (p = 0,002).
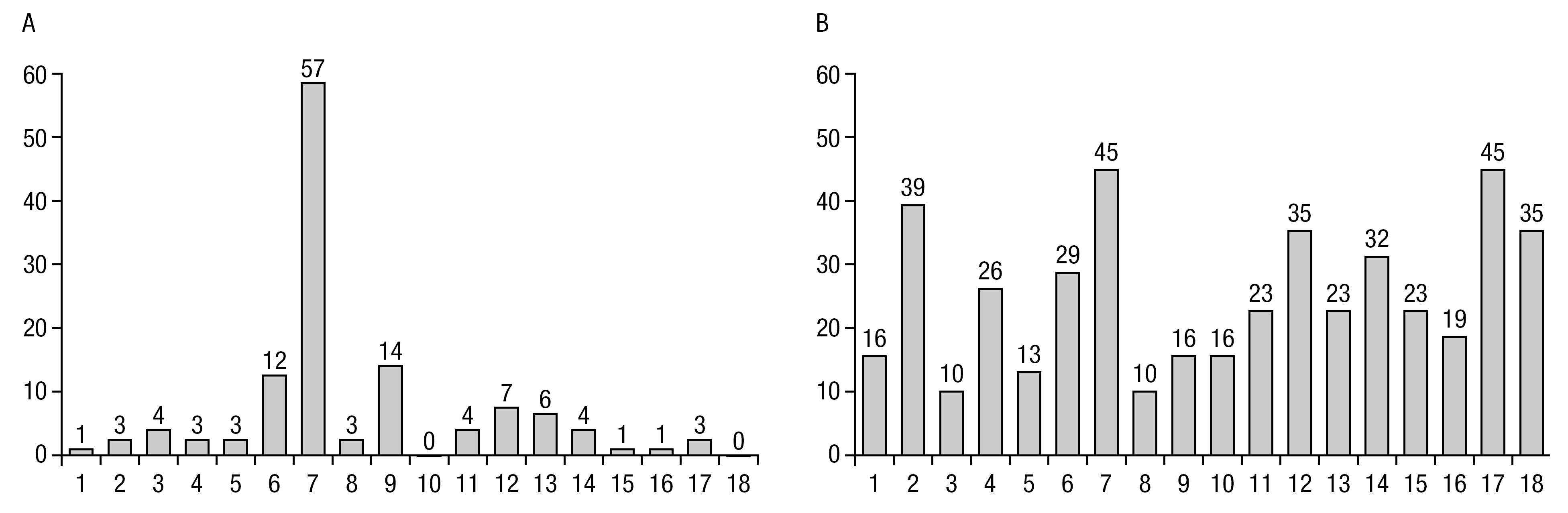
Em relação à análise comparativa das frequências percentuais de não metilação por cada CpG, verificaram-se diferenças significativas (p = 0,000-0,044) entre a normozoospermia e a oligozoospermia grave em todos os casos, com excepção dos locais CpG-3, 5, 7, 8 e 9. Em relação à normozoospermia, a percentagem de não metilação foi sempre inferior a 10%, sendo as citosinas com maior percentagem de desmetilação as CpG-6 e 9 (fig. 4A). Pelo contrário, na oligozoospermia severa, a percentagem de não metilação foi praticamente sempre superior a 10%, encontrando-se as maiores percentagens de não metilação (> 30% dos clones) nas CpG-2, 12, 14, 17 e 18 (fig. 4B). Em duas CpGs de amostras com normozoospermia, as CG-10 e CG-18, todos clones se encontraram totalmente metilados, enquanto que nos clones da oligozoospermia severa estas CG apresentaram uma percentagem de não metilação de 16% e 35%, respectivamente.

Figura 4. Frequências percentuais (eixo dos Y) de clones com défice de metilação na (A) normozoospermia (n = 69) e na (B) oligozoospermia severa (n = 31) por cada CG (eixo das abcissas). O local da CpG-7 é polimórfico.
DISCUSSÃO
Os resultados obtidos revelam que os pacientes com oligozoospermia grave apresentam defeitos na metilação do gene H19, traduzidos pelo elevado número de citosinas que se apresentavam não metiladas neste gene (83,9%) e pela elevada frequência de múltiplas CpG não metiladas em simultâneo, quer fora quer dentro da região de ligação da CTCF. O facto de nestes pacientes se terem encontrado dois clones totalmente não metilados sugere um risco acrescido de transmissão de defeitos de imprinting, uma vez que o alelo paterno deste gene é metilado nos espermatozóides. Adicionalmente, em 3 dos clones de oligozoospermia grave verificou-se uma ausência total de metilação em todas as cinco CG que constituem o local de ligação da proteína reguladora CTCF6 (CpG 4-8). Este tipo de erro de imprinting nos espermatozóides poderá conduzir a um desenvolvimento anormal do embrião, uma vez que o défice da metilação activa o gene H19 (pelo que dois genes H19 activos estarão presentes no embrião) e inibe o gene IGF2 (pelo que dois genes IGF2 inactivos estarão presentes no embrião)24-26.
Os espermatozóides de homens normozoospérmicos apresentaram também uma metilação incompleta das citosinas, embora em muito menor número. Este facto sugere que será mais importante na transmissão de um imprinting correcto o grau de metilação global do gene e não a metilação de algumas citosinas em particular. No entanto, verificou-se uma diferença significativa na ocorrência de não metilação na maioria das CpGs, sugerindo que estas CpGs podem estar mais susceptíveis à desmetilação nos espermatozóides de oligozoospérmicos graves.
A ausência de metilação nas CpG do gene H19 pode ser devida a dois factores, ou por défices do processo de desmetilação fetal, ou por défice de metilação diferencial pelas DNA-metiltransferases, enzimas que estabelecem a metilação dos genes imprinted durante a gametogénese. Os nossos resultados prévios da análise do gene MEST21 sugerem que a não metilação não se deve a défices do processo de desmetilação global. De facto, o gene MEST, um gene que se mantém desmetilado na espermatogénese, encontrou-se sempre não metilado nas amostras de oligozoospermia grave. Estes resultados sugerem, assim, que na oligozoospermia grave existe uma actividade anómala das DNA-metiltransferases, o que as implica pela primeira vez nos defeitos na espermatogénese.
Dois estudos prévios haviam também estudado em espermatozóides a região hcs-6 da DMR do gene H19. Num desses trabalhos, os autores verificaram uma metilação completa das CG, tendo analisado 30 clones a partir de três ejaculados com concentração normal de espermatozóides4. Pelo contrário, os nossos resultado demonstram um défice de metilação de 47,8% nas amostras oriundas de ejaculados com concentração normal de espermatozóides, sendo esta diferença explicável pelo maior número de amostras que estudamos. No outro estudo, efectuado em 16 clones oriundos de espermatozóides de um ejaculado com concentração normal de espermatozóides, os autores verificaram que podem existir 1-3 CpGs não metiladas, tendo sido a CG-14 a que apresentava maior percentagem de não metilação (9/16, 56%)3. Estes resultados são parecidos com os do nosso estudo, pois também encontramos 1-3 CG não metiladas na normozoospermia, mas diferem totalmente nas frequências, pois verificamos que os níveis de não metilação raramente ultrapassam os 10% por cada CpG, com excepção da CG-6 (12%) e da CG-9 (14%).
Um outro trabalho prévio estudou o nível de metilação do gene H19 em espermatozóides de homens normozoospérmicos, mas em regiões distintas da hcs6, e que incluíram as regiões hcs-1, hcs-3 e hcs-5. A região hcs-1 apresentou desmetilação parcial, a região hcs-3 encontrou-se completamente desmetilada em todos os clones, e a região hcs-5 estava totalmente metilada em quase todos os clones, o que sugere a importância das regiões hcs-1 e hcs-5, mas não da hcs-3, no estabelecimento da marca de imprinting do gene H195.
Os estudos efectuados na DMR do gene H19 em espermatozóides de rato27-29, confirmam os estudos prévios em espermatozóides humanos, uma vez que, e apesar do reduzido número de clones avaliado, a maioria dos casos se encontrava totalmente metilada, enquanto que nas com metilação incompleta esta apenas variava entre 1-3 CpGs não metiladas. No entanto, os dados do nosso estudo mais alargado demonstram uma diferença significativa entre as duas espécies.
Em conclusão, efectuamos um estudo alargado da metilação da região hcs-6 da DMR do gene H19 em espermatozóides oriundos quer de ejaculados com concentração normal de espermatozóides (69 clones), que de amostras com oligozoospermia grave (31 clones). Este estudo é o primeiro a ser efectuado numa amostra tão ampla de clones nos casos com concentração normal de espermatozóides, e a primeira a ser efectuada em casos de oligozoospermia grave. Os resultados demonstram que, na normozoospermia, podem haver CG não metiladas, mas que a sua frequência raramente atinge os 10% e não ultrapassa 3 CG não metiladas em simultâneo (5/33, 15,2%). Pelo contrário, na oligozoospermia grave, a maioria dos
espermatozóides apresenta défices de metilação (83,9%), a maioria das CpG é atingida com frequências superiores a 20% (11/26, 42,3%), na maior parte dos casos ocorrem défices de metilação em várias CG em simultâneo (19/26, 73,1%), e ocorrem casos com não metilação de toda a região (2/31, 6,5%), bem como casos com não metilação de todas as CG envolvidas na ligação da CTCF (3/31, 9,7%). Por ficar assim demonstrado um aumento significativo de risco de transmissão de defeitos do imprinting genómico paterno na oligozoospermia, sugere-se que antes do tratamento da infertilidade por ICSI se peça um estudo do imprinting genómico dos espermatozóides para avaliação do risco.
AGRADECIMENTOS
Agradece-se o apoio laboratorial nos espermogramas do Dr. Paulo Viana (Centro de Genética da Reprodução A. Barros).
O presente trabalho foi parcialmente financiado pela Fundação para a Ciência e Tecnologia (Sapiens: 36363/99, 43462/01, 48.376/02, UMIB).
Correspondencia: Mário Sousa.
Lab Biologia Celular. Instituto de Ciências Biomédicas de Abel Salazar.
Universidade do Porto.
Lg Prof Abel Salazar, 2. 4099-003 Porto. Portugal.
Correo electrónico: msousa@icbas.up.pt